Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Dicembre 2004 - Volume VII - numero 11
M&B Pagine Elettroniche
Pediatria per l'ospedale
Emofilia
A e B
1°
parte
Le
emofilie sono malattie ereditarie nelle quali viene a mancare uno dei
fattori della coagulazione: le più conosciute sono la emofilia
e la emofilia B, anche se la malattia di Willebrand è la più
frequente. I notevoli miglioramenti nella diagnosi e soprattutto lo
sviluppo di trattamenti sicuri ed efficaci, hanno permesso che i
soggetti che ne siano affetti, possano condurre una vita normale.
Rimangono da risolvere ancora problemi di grande importanza, come la
comparsa di complicazioni, particolarmente lo sviluppo di anticorpi
verso il fattore impiegato, che pertanto diviene inefficace, o la
comparsa di infezioni, trasmesse dal sangue o dai suoi derivati. Il
seminario di Bolton-Maggs PHB e Pasi KJ ci offre un aggiornamento
interessante e completo (Haemophilias A and B, Lancet 2003,
361:1801-09).
Le
emofilie sono malattie ereditarie della coagulazione caratterizzate
dalla bassa concentrazione di specifici fattori della coagulazione.
Le forme più conosciute sono quelle dovute a mancanza del
fattore VIII (emofilia A) e del fattore IX (emofilia
B), ambedue dovute ad alterazioni geniche X-legate. La mancanza
del fattore XI (originariamente chiamata emofilia C) è meno
comune e in molti casi si accompagna ad alterazioni lievi della
coagulazione, ereditate come carattere autosomico recessivo,
particolarmente comune negli ebrei Ashkenazi.
Tuttavia
la più comune malattia ereditaria è la malattia di von
Willebrand, dovuta a un'alterazione quantitiva e qualitativa del
fattore di von Willebrand, presente in quasi l'1% della
popolazione in generale. Questa malattia è generalmente lieve,
ma può rappresentare un'importante causa di menorragia nelle
famiglie colpite. Va sottolineato il fatto che la diagnosi di questa
affezione non è affatto esclusa dal reperto di normali esami
della coagulazione.
Deficienze
congenite di altri fattori della coagulazione sono molto rare: si
tratta di condizioni autosomico recessive, più frequenti nelle
comunità nelle quali sia comune la consanguineità.
L'emofilia
A e B sono clinicamente indistinguibili l'una dall'altra. La
diagnosi e quindi il riconoscimento dell'una o dell'altra vanno
confermati con la ricerca del fattore specifico. La tendenza al
sanguinamento è in relazione alla concentrazione del fattore
ed è classificata come lieve, moderata e grave (vedi
tabella 1). Questa classificazione valuta il rischio di
sanguinamento, indica il trattamento migliore e predice l'evoluzione.
Mentre la maggioranza dei pazienti con le forme gravi di emofilia
necessita di un trattamento regolare di sostituzione, pochi di loro
sanguinano spontaneamente e necessitano quindi di un trattamento
occasionale. Va ricordato che l'eredità anche di geni
trombofilici può modificare l'espressione clinica
dell'affezione.
I difetti
congeniti dei fattori della coagulazione che richiedano un
trattamento specialistico sono rari (vedi
tabella 2). Nel Regno Unito vi sono circa 5.000 soggetti con
emofilia A; la prevalenza dell'emofilia B è circa un quinto
di quella dell'emofilia A. A livello mondiale viene calcolato che
vi siano mezzo milione di persone con emofilia A, con una prevalenza
fra 105 e 160 per milione di popolazione maschile.
Fisiopatologia
Il
fattore VIII è una glicoproteina plasmatica complessa di 2.351
aminoacidi, sintetizzata principalmente dagli epatociti, sebbene il
rene, le cellule sinusoidale endoteliali e i tessuti linfatici
possano sintetizzarla in minima quantità. Si tratta di uno dei
più grandi e meno stabili fattori della coagulazione,
circolanti nel plasma in un complesso non covalente con il fattore di
von Willebrand. Il fattore VIII ha un'emivita di 12 ore negli
adulti e un po' meno nei bambini. Il fattore di von Willebrand
protegge il fattore VIII dalla degradazione proteolitica prematura e
lo concentra a livello della lesione vascolare.
Il
fattore IX è una proteasi sierica di 415 aminoacidi,
sintetizzata a livello del fegato: esso è la più grande
proteina K-dipendente. La vitamina K è necessaria per
permettere la gamma carbossilazione terminale dei residui di acido
glutammico per formare i domini Gla, che sono essenziali per il
normale funzionamento e quindi per l'attività biolologica.
La concentrazione plasmatica del fattore IX è circa 50 volte
quella del fattore VIII; il fattor IX ha una metà vita di
circa 24 ore.
Concentrazioni
del fattore VIII e IX | Classificazione | Clinica |
<0,01
UI/mL (<1% del normale) | Grave | Sanguinamenti
spontanei delle articolazioni e dei muscoli; sanguinamenti dopo
traumi, incidenti e interventi chirurgici. |
0,01-0,05
UI/mL (1-5% del normale) | Moderate | Sanguinamenti
nelle articolazioni e nei muscoli in seguito a minimi traumi;
eccessivi sanguinamenti dopo interventi chirurgici ed estrazioni
dentarie |
>0,05-0,4
UI/mL (5-40% del normale) | Lieve | Non
avvengono sanguinamenti spontanei, sanguinamenti dopo interventi
chirurgici, estrazioni dentarie e incidenti. |
Tabella
2 – Prevalenza delle mancanza di fattori rari della coagulazione in
confronto a quella della emofilia A e B
Mancanza
del fattore | Prevalenza
stimata della deficienza grave (x 106)* | Gene
sui cromosomi |
Fattore
VIII o fattore IX | 133
nel 106maschi** | X |
Fibrinogeno | 1 | 4 |
Protrombina | 0.5 | 11 |
Fattore
V | 1 | 1 |
Combinato
V e VIII | 1 | 18 |
Fattore
VII | 2 | 13 |
Fattore
X | 10 | 13 |
Fattore
XI | 1*** | 4 |
Fattore
XIII | 0.5 | 6
(subunità A)
1
(subunità B) |
*Concentrazione
del fattore <10% del normale.
**Dati
combinati dei fattori VIII e IX con tutte le gravità.
***Particolarmente
negli Ebrei Ashkenazi, fra i quali la stima della prevalenza della
mancanza grave è di 1 su 190 e l'8,1% della popolazione è
eterozigote.
L'emostasi
e il ruolo dei fattori VIII e IX
Il
sanguinamento avviene nell'emofilia a causa dell'insufficienza
dell'emostasi secondaria. L'emostasi primaria, la formazione del
tappo piastrinico avvengono normalmente, ma la stabilizzazione del
tappo di fibrina è difettoso per l'inadeguata quantità
di trombina prodotta.
Sebbene
la classica ipotesi della cascata della coagulazione proponga due vie
separate, i fattori VIII e IX rappresentano il punto centrale del
processo di coagulazione e per l'adeguata produzione della trombina
(Vedi
figura 1).
Dopo la
lesione, l'attivazione del complesso del fattore tissutale e del
fattore VII media la produzione del fattore Xa. Questa produzione
deve essere amplificata dal fattore VIII e IX che permettono alla
coagulazione di progredire verso il completamento. E' evidente che
in assenza del fattore VIII e IX, l'amplificazione e la generazione
consolidata del fattore Xa è insufficiente per mantenere
l'emostasi.
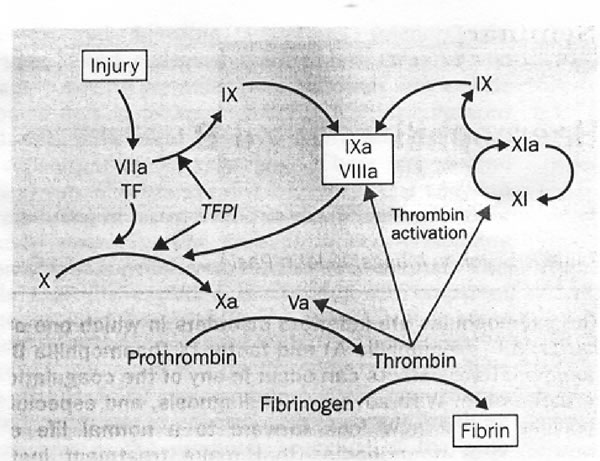
La
coagulazione inizia quando il danno del tessuto espone il fattore
tissutale (TF). Il fattore VII si lega al fattore cellulare; il
complesso attiva direttamente il fattore X a Xa e parte del fattore
IX a fattore IXa. In presenza del fattore Xa, l'inibitore del
fattore tissutale (TPFI) inibisce l'ulteriore produzione del
fattore Xa e IXa. Dopo questa inibizione la quantità di
fattore Xa prodotta non basta a mantenere la coagulazione.
L'ulteriore produzione di fattore Xa per permettere all'emostasi
di completarsi, può derivare solo dal fattore VIII-IX. Viene
così formata sufficiente trombina per attivare il fattore
VIIIa e insieme il fattore IXa, che attivano ulteriori quantità
di fattore X. Un aumento dell'attivazione del fattore del IX
avviene attraverso la via del fattore XI.
Genetica
molecolare dell'emofilia
I geni
del fattore IX e VIII sono stati clonati trispettivamente nel 1982 e
nel 1984. Questa clonazione è stata determinante per la
produzione dei concentrati di fattori della coagulazione
ricombinanti, per uso terapeutico, per la generazione di animali
knockout da usare come modelli di emofilia e infine per la produzione
di proteine di tipo normale e di mutanti per l'analisi del rapporto
funzione/struttura delle proteine.
Emofilia
B: mutazioni del fattore IX
Il
fattore IX contiene 8 esoni e misura 33,5 kb. Esso si trova sul
braccio lungo del cromosoma X, all'Xq27. Il gene del fattore IX è
molto più piccolo e meno complesso del gene del fattore VIII.
Sono
state riscontrate più di 2.100 mutazioni del gene del fattore
IX: esse sono state descritte in tutte le regioni del gene; nella
maggioranza dei casi si tratta di mutazioni puntiformi, spesso (2/3)
di tipo missense. Circa il 20-30% dei casi di emofilia B è
dovuto a un piccolo numero di mutazioni “founder”. Circa il 7%
sono piccole aggiunte o delezioni e il 3% è rappresentato da
mutazioni grossolane o da riarrangiamenti complessi. La sostituzione
nella regione promotrice del fattore IX classicamente risulta nel
raro fenotipo fattore IX Leiden.
Emofilia
A: mutazioni del gene del fattore VIII
Il
gene del fattore VIII è formato da 186 kb, consiste di 26
esoni e si ritrova sul braccio lungo del cromosoma X a Xqw28. Esso ha
all'interno dell'introne 22, due geni addizionali (F8A ed F8B).
Due copie del fene F8A si ritrovano al di fuori del gene del fattore
VIII (400 kb telomerica). La funzione di questi due geni addizionali
non è conosciuta.
Il più
comune difetto genetico nell'emofilia A, che colpisce il r45% dei
soggetti con malattia grave, ha una larga inversione e traslocazione
degli esoni 1-22 (insieme con gli introni). A parte questa mutazione
le altre sono quasi sempre delle mutazioni puntiformi (circa 85%
mis-sense e il 15% non-sense), di cui il 5% sono grandi o piccole
delezioni o inserzioni.
Rischio
di comparsa degli inibitori e difetto molecolare
Il
rischio di comparsa degli inibitori è associato al tipo di
mutazione presente. Nella emofilia A i pazienti con mutazione che
tronca in modo grave o previene la produzione del fattore VIII
(inversione dell'introne 22, grandi delezioni, mutazioni non-sense)
hanno una frequenza più elevata (circa il 35%) dello sviluppo
di inibitori di quelli che hanno una mutazione mis-sense o piccole
delezioni (circa il 5%), nelle quali una piccola quantità
della proteina può essere prodotta.
Nella
emofilia B i pazienti con delezione del gene o con riarrangiamenti
hanno un rischio di sviluppare inibitori di circa il 50%, mentre per
mutazioni “frameshift”, stop prematuri o mutazioni “splice-site”
il rischio è di circa il 20%. Per quelli che hanno mutazioni
mis-sense il rischio è quasi uguale a zero.
Diagnosi
L'emofilia
viene diagnosticata:
- In base alla storia familiare (che manca in un terzo degli emofilici)
- O in base alla presenza di un'emorragia.
La
maggior parte dei bambini nasce senza complicazioni in seguito a un
parto vaginale. La stima dell'emorragia intracranica nel periodo
neonatale è fra l'1 e il 4%: questa complicazione è
più facile entro la prima settimana di vita. L'emorragia è
particolarmente a rischio quando si usi il vacuum extractor. Quando
la storia della famiglia fa prevedere la possibilità della
nascita di un bambino ammalato di emofilia, è essenziale una
buona comunicazione fra l'ostetrico, il pediatra e l'ematologo.
La maggioranza dei bambini è senza sintomi finchè non
comincino a gattonare o a camminare.
Il punto
essenziale dell'emofilia grave è la comparsa spontanea di
sanguinamenti nelle articolazioni e nei muscoli, che portano alla
comparsa di dolore e di lesioni distruttive se non adeguatamente
trattati. La maggioranza dei bambini con grave emofilia presenta il
suo primo sanguinamento in un'articolazione dall'età di 4
anni in poi, ma sanguinamenti in altre sedi sono già presenti
prima di questa età. I lattanti colpiti sanguinano facilmente
e il sospetto di una lesione non accidentale può sorgere nel
trattare precocemente una lesione traumatica.
L'emofilia
moderata viene diagnosticata nella maggioranza dei casi dopo l'età
di 5 anni, anche se a volte questa emofilia viene diagnosticata più
tardi nella vita dopo un trauma o un intervento chirurgico.
Va
ricordato che la concentrazione di fattore VIII non cambia in modo
significativo con l'età nella emofilia A.
La
deficienza del fattore VIIIC, dovuta all'emofilia A deve essere
distinta dalla malattia di von Willebrand dosando il fattore von
Willebrand. La storia familiare (con la trasmissione autosomica) e i
sintomi della emorragia (menorragia, facile comparsa di ematomi ed
epistassi) aiutano nella diagnosi differenziale. La maggior parte dei
casi di malattia di von Willebrand sono lievi; emartri e
sanguinamenti muscolari insorgono soltanto nella forma grave tipo 3
(molto rara) nella quale il fattore di von Willebrand è
assente e la concentrazione di fattore VIIIC è molto bassa. Ci
sono molti sottotipi di malattia di von Willebrand, incluso uno
dovuto alla difettosa unione del fattore VIIIC al fattore di von
Willebrand (variante Normandy). La quantità di fattore di von
Willebrand è normale in questi casi, ma è la
combinazione con il fattore VIII che è anormale. Questa
diagnosi deve essere esclusa nelle famiglie con apparente emofilia A
lieve o moderata, specialmente se alcune donne della famiglia hanno
una concentrazione bassa di fattore VIIIC.
Il
dosaggio del fattore IX conferma la diagnosi di emofilia B. La
concentrazione del fattore IXC non cambia significativamente con
l'età nei soggetti colpiti, eccetto per quelli con mutazione
nella sequenza del gene (fattore IX Leiden), nella quale la
concentrazione aumenta con le modificazioni ormonali alla pubertà.
La grave emofilia B diviene lieve e i pazienti con emofilia B lieve
possono sviluppare una normale concentrazione di fattore IXC.
L'identificazione di queste famiglie è importante perché
la prognosi è migliore.
Le
emofilie A e B sono malattie legate al sesso; poiché la
concentrazione media del fattore nei portatori è circa la metà
del normale, una buona parte delle donne portatrici ha basse
concentrazione dei fattori VIII C e IXC, che possono predisporre a un
eccesso di emorragie. La concentrazione dei fattori deve essere
misurata nelle ragazze e nelle donne che sono delle sicure o delle
possibili portatrici. La concentrazione del fattore VIIIC deve essere
misurata più di una volta, perché la concentrazione può
aumentare sotto stress, essendo il fattore VIII una proteina della
fase acuta. Anche concentrazioni normali, riscontrate più
volte non escludono uno stato di portatore, che può essere
determinato con sicurezza solo dall'identificazione della
mutazione. Questa prova genetica può essere eseguita soltanto
quando la ragazza abbia raggiunto l'età per consentirlo (fra
12 e 16 anni).
Raramente
le donne hanno una grave emofilia: questa può verificarsi
quando:
- C'è un estrema lionizzazione
- Nella sindrome di Turner
- Quando ambedue i genitori siano portatore del gene patologico (padre con emofilia e madre portatrice)
Quando in
una famiglia venga posta la diagnosi di emofilia, il clinico deve
raccogliere un'accurata storia familiare e deve costruire un
accurato pedigree.
Vuoi citare questo contributo?