Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Aprile 1999 - Volume II - numero 4
M&B Pagine Elettroniche
Contributi Originali - Ricerca
Sindrome
di Waardenburg e sindrome del primo arco. Uno studio genealogico
Mario
Lagrasta
Pediatra
di Base, ASL Ba , Corato, Bari
Waardenburg's
syndrome and first arch syndrome. A genealogical study
Aim of
the Work
Answering
the following questions: a) Is iris heterochromia a monosymptomatic
form of Waardenburg's syndrome? b) May deaf-mutism and cleft lip
have the same genetic substrate? c) Are these anomalies due to a
monogenic defect?
Materials
and Methods
9
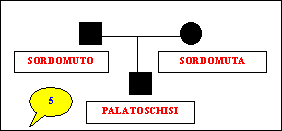
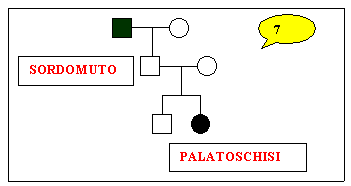
genealogical trees were studied. For 7 families, cases of deaf-mutism
and cleft lip (or cleft palate, or palate agenesis, or Pierre-Robin's
syndrome) were found in two or three different generations; whereas
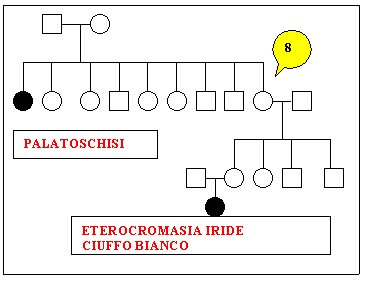
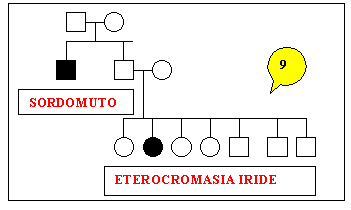
for 2 other families, cases of cleft palate and iris heterochromia
with or without white tuft were found (Waardenburg's syndrome).
All these
cases can be classified under a single definition of first arch
syndrome (McKenzie), whose expressions can be very different (Havel).
All these anomalies are related to abnormal face structure
developments to be regarded as different expressions of a single
development defect (mandibular-facial dysostosis or Teacher-Collins'
syndrome, Pierre Robin's syndrome, deformation of external and
middle ear, deaf-mutism, cleft lip and cleft palate, hypertelorism
and, last but not least, congenital deafness with hypertelorism,
white tuft and iris dyschromia or Waardenburg's syndrome). A
genetic defect (Hox gene) is responsible for the malformation or the
absence of stapes artery. Hence, the absence of vascularisation is
responsible for one of the expressions of the first arch syndrome.
Such an expression and its penetration depend upon the rate of
development of collateral cephalus.
Conclusions
These
remarks do confirm the assumption that the aforementioned
malformations are not due to accidental embryogenetic mistakes, but
to a single genetically controlled defect.
recent
survey has shown an increase in exposure (10%) and expenditure (12%)
for antibiotics. New active substances are preferred. This research
is aimed at assessing this aspect of drug-epidemiology with reference
to the age group 0-14.
Articolo
Premesse
e scopo del lavoro
Obiettivi:
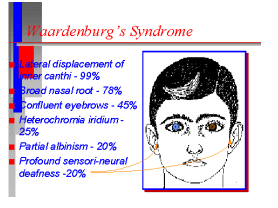
1)
l'eterocromasia dell'iride rappresenta la forma monosintomatica
della sindrome del primo arco o meglio della sindrome di
Waardenburg?
2) un
sordomuto e un labbro leporino o palatoschisi hanno lo stesso
genotipo?
3) tali
malformazioni sono dovute a difetti geneticamente controllati?
Materiali
e metodi
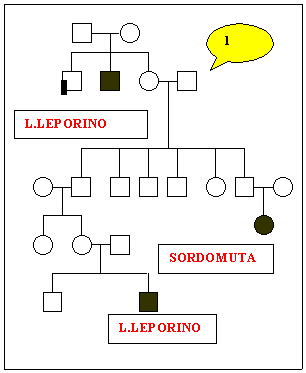
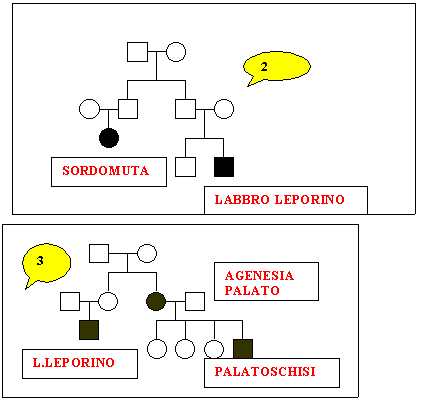
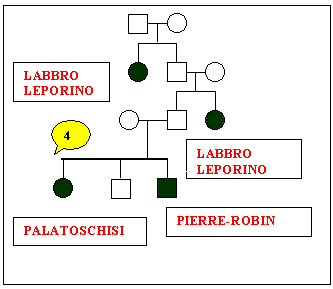
Sono
stati studiati nove alberi genealogici che sembrano confortare i
nostri obiettivi.

Conclusioni
Tutte
queste manifestazioni possono raggrupparsi sotto il termine unico di
sindrome del primo arco (McKenzie).
Si tratta
d'anomalie di sviluppo delle ossa facciali da considerare comemanifestazioni diverse di un unico errore di sviluppo.
Hovels
riscontrò diversi gradi del processo evolutivo della malattiadalla semplice obliquità delle fessure oculari fino
alle più gravi ipoplasie facciali.
-Sindrome
di Treacher –Collins (disostosi mandibolo facciale)
-Sindrome
di Pierre –Robin
-Deformità
dell'orecchio esterno e medio
-Sordomutismo
congenito
-Labbro
leporino e palatoschisi
-Ipertelorismo
-Sordità
congenita, ipertelorismo con ciuffo bianco, occhi di differente
colore, ciglia bianche (sindrome di Waardenburg)
"L'origine
di queste anomalie sembra dovuto ad un difetto di
vascolarizzazione del primo arco in via di sviluppo. L'errore
primitivo sta nella precoce involuzione dell'arteria della staffa.
La gravità dell'anomalia che ne deriva dipende dalla
velocità con la quale s'instaura un circolo collaterale.
Tali anomalie si trasmettono con carattere dominante a penetranza
variabile".(Schaffer)
Il
difetto genetico (un gene hox) è responsabile della
malformazione o assenza dell'arteria della staffa, mentre il
circolo collaterale rende conto del grado di penetranza.
La
penetranza esprime la relazione tra genotipo e fenotipo: quando la
penetranza è completa, il 100% degli individui che portano un
determinato genotipo mostrano lo stesso fenotipo.
Nella
sindrome del primo arco la penetranza è incompleta in quanto
solo una certa proporzione degli individui che portano un determinato
genotipo manifesta il fenotipo corrispondente, mentre gli altri
individui sono normali: ad uno stesso genotipo corrisponde un
fenotipo diverso.
Questo
spiega i salti di generazione che si traduce in un'irregolarità
nella trasmissione verticale tipica dei caratteri o malattie
autosomiche dominanti.
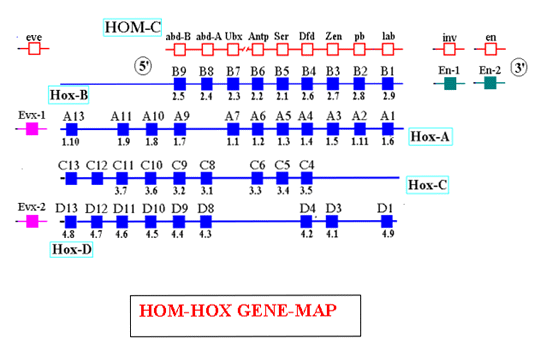
I geni
hox fanno parte di un gruppo di geni altamente conservati che
codificano –quando non mutati- per il limite dello sviluppo.
I geni hox sono geni omeotici cioè geni mutanti il cui effetto
sul fenotipo è l'omeosi in altre parole la trasformazione di
singole strutture nelle sembianze di altre anteriori o posteriori
rispetto alla normalità.
La mappa
dei geni Hox è composta da quattro complessi A, B, C, D
derivanti da due successive duplicazioni geniche di un cluster
ancestrale, avvenute in due occasioni diverse.
In
seguito a queste duplicazioni geni ubicati in complessi diversi ma in
posizioni relativamente identiche sono più simili rispetto a
geni localizzati nello stesso cluster: geni paralogues. Oltre
l'omeosi i geni hox influenzano gli schemi di crescita: geni
architetti.
“Essi
provvedono alla modellizzazione del corpo e regolano la sequenza di
formazione del capo e degli arti in senso cranio caudale e
prossimo-distale.
Questo
tipo d'osservazioni ci dice che le malformazioni suddette non sono
dovute ad errori accidentali nell'embriogenesi ma a difetti
geneticamente controllati.”(MeB dic. '97)
Bibliografia
Schaffer
AJ:"Sindrome del primo arco" in Le Malattie Del
Neonato, Vol. II, pag. 807, Piccin, Padova,1970.
Panizon
F: Novità in pediatria pratica 1996-1997, Medico e
Bambino: 10, 45, 1997
Jeffrey
W.Innis: Ruolo dei geni hox nello sviluppo umano. Current Opinion
in Pediatrics. Ed.Italiana 1,1,52, 1998
Vuoi citare questo contributo?