Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Aprile 2009 - Volume XII - numero 4
M&B Pagine Elettroniche
Pediatria per immagini
Ali
di farfalla
Clinica
pediatrica, IRCCS Burlo Garofolo, Trieste
Indirizzo
per corrispondenza: rizzello.elisa@libero.it
D. è
una bambina di 2 anni con cardiopatia su base malformativa
(aneurisma del setto interatriale e pervietà del forame ovale)
diagnosticata alla nascita, del tutto asintomatica. Fin dai primi
mesi di vita la mamma riferisce che la piccola vomita dopo i pasti ed
ha un prurito importante che persiste nonostante il trattamento con
antistaminici, rifampicina e acido ursodesossicolico. Pertanto si
decide di indagare il problema, e le indagini di laboratorio rilevano
uniperlipidemia (colesterolo totale 473 mg/dl; v.n. < 180
mg/dl, trigliceridi 236 mg/dl; v.n. 30-100 mg/dl) associata ad un
moderato aumento di bilirubina diretta 0,66 mg/dl (v.n. < 0,5
mg/dl), AST (206 U/l; v.n. 8-20 U/l), ALT (139 U/l; v.n. 8-20 U/l),
γ-GT (506 U/l; v.n. 4-90 U/l) e fosfatasi alcalina (2.082 U/l;
v.n. 110-550 U/l). Nel sospetto di unepatopatia colestatica si
esegue unecografia epatica, risultata nella norma; inoltre,
per valutare levoluzione della cardiopatia di cui è
affetta viene effettuata un ecocardiografia che rileva una
moderata stenosi periferica dellarteria polmonare. Pensando ad
un quadro sindromico, infine, viene valutata la presenza di eventuali
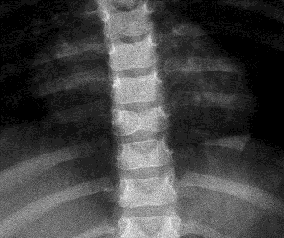
alterazioni ossee mediante un Rx torace che, difatti, mostra una
fusione dellarco vertebrale anteriore di T8, conosciuto come
vertebra a farfalla (Figura 1). Tutti i segni
e sintomi descritti hanno quindi suggerito la diagnosi di sindrome
di Alagille, in seguito confermata da indagini genetiche.
La
sindrome di Alagille
La
sindrome di Alagille è un disturbo multisistemico che
colpisce 1/70.000 nati vivi ogni anno; la sua diagnosi si basa sui
criteri clinici stabiliti da Alagille nel 1975, i quali includono
paucità dei dotti biliari alla biopsia epatica associata ad
almeno tre delle cinque principali manifestazioni cliniche: colestasi
cronica, difetti cardiaci congeniti, anomalie vertebrali, anomalie
oculari e facies caratteristica6 (Tabella
I). La maggior parte dei casi sono incompleti o mild,
con unaspettativa di vita pressochè normale, e bassa
mortalità in età pediatrica; le forme con espressività
completa hanno, al contrario, una prognosi peggiore5. La
malattia è provocata da mutazioni (riarrangiamenti e
delezioni) del gene JAGGED1 localizzato sul braccio corto del
cromosoma 20; esso codifica per un ligando del recettore
transmembrana Notch, il cui segnale media i meccanismi di
differenziazione cellulare; lereditarietà è
autosomica dominante, con penetranza ed espressività
variabili, determinando uno spettro di severità clinica molto
ampio3-4-5.
La
colestasi è presente nella quasi totalità dei
pazienti; si manifesta solitamente nei prima 2 anni di vita con la
comparsa di ittero, epatosplenomegalia, feci acoliche, urine
ipercromiche e malassorbimento, associati ad un aumento degli indici
di colestasi (bilirubina diretta, sali biliari, transaminasi, γ-GT,
fosfatasi alcalina) e iperlipidemia (colesterolo totale e
trigliceridi). La metà dei casi evolve in ipertensione
portale7. A tutto ciò, nell80% dei casi, si
associa scarsa crescita legata ad un bilancio energetico negativo
secondario ad incremento del consumo energetico, ridotto introito
calorico e malassorbimento lipidico; i bambini con sindrome di
Alalgille, infatti, presentano il difetto di crescita più
severo tra tutte le forme di colestasi12. La diagnosi,
oltre che clinica e laboratoristica, utilizza tecniche di imaging
(ultrasonografia, colangiografia, colescintigrafia, colangio-RMN), le
quali rilevano latresia biliare con riduzione della secrezione
nel tratto gastrointestinale (figura 2 e
Figura 3). La conferma diagnostica è
data dalla biopsia epatica e dalla ricerca delle mutazioni
genetiche7.
Il
prurito intenso è spesso presente, ed è
solitamente il sintomo più debilitante; è causa di
lesioni da grattamento, escoriazioni, lichenificazioni fino a vere e
proprie mutilazioni cutanee, oltre che insonnia, difficoltà di
attenzione e riduzione del rendimento scolastico e, occasionalmente,
suicidio9. Unaltra manifestazione cutanea,
secondaria a colestasi protratta e severa, è la formazione di
xantomi (28-42% dei casi); la localizzazione più comune
è la superficie estensoria delle dita, seguita da solchi
palmari, nuca, gomiti, ginocchia, zona glutea e perianale, fossa
poplitea e inguine. Essi compaiono progressivamente a partire dai 4
anni anni, per poi regredire dopo i 10 anni detà.
Unaltra caratteristica cutanea consiste nella comparsa di
pieghe flessorie digitali sovrannumerarie (35%), localizzate
soprattutto sulle falangi intermedie di uno o più dita; meno
comuni sono invece linfedema delle estremità, eritema palmare,
xerosi, ipercheratosi follicolare e teleangectasie8.
I difetti
cardiaci congeniti sono un altro criterio diagnostico principale
della sindrome di Alagille; il più comune è
rappresentato dalla stenosi polmonare periferica (85%), anche
se sono riportati casi di stenosi centrale. Altre anomalie descritte
sono difetti di setto interatriale e interventricolare, dotto
arterioso pervio e coartazione dellaorta5.
Le
anomalie vertebrali son le più frequenti alterazioni
scheletriche riscontrate in questi pazienti; colpiscono il 66% dei
soggetti affetti, il 48% dei quali presenta un interessamento
vertebrale multiplo. La vertebra a farfalla è quella
maggiormente riscontrata (alcuni autori le conferiscono una
prevalenza variabile tra 50%-80%); essa è secondaria ad una
mancata fusione, completa o incompleta, dellarco vertebrale
anteriore (più spesso individuata a livello T6-T9). Nella
maggior parte dei casi è simmetrica, e quindi clinicamente
muta rappresentando un reperto isolato non sempre semplice da
identificare con indagini radiologiche1-2; le rare forme
sintomatiche sono quelle asimmetriche, le quali possono presentarsi
con scoliosi di grado variabile. Le anomalie dello scheletro
appendicolare sono invece meno frequenti; tra queste troviamo
ipoplasia delle falangi distali delle dita (16%), accorciamento
ulnare (13%) ed anomalie di coste e ossa pelviche (5-8%)4.
Lembriotoxon
posteriore è lalterazione oculare più
frequentemente descritta nei pazienti con sindrome di Alagille
(78-95% dei casi); esso risulta dalla persistenza di residui
embrionali e dal dislocamento anteriore della linea di Schwalbe
nellangolo sclero-corneale, facilmente diagnosticato con
lampada a fessura. Altre alterazioni sono anomalie della
pigmentazione di iride e retina, microcornea, alterazioni del fondo
oculare (papilla, vascolarizzazione), drusen del nervo ottico;
lacuità visiva è solitamente conservata10.
La facies
in età pediatrica è caratterizzata da fronte
prominente, occhi infossati con moderato ipertelorismo, radice nasale
profonda con cresta nasale dritta, e mento prominente e appuntito;
tutte queste peculiarità conferiscono al viso la forma di un
triangolo invertito, rappresentando un criterio diagnostico
rilevante, molto sensibile e specifico (Figura 4).
Tale fenotipo faciale è presente infatti nel 95% dei pazienti.
In epoca post-puberale e adulta la facies si modifica, cosicchè
il mento diviene sempre più prominente fino ad un vero e
proprio prognatismo, mentre la fronte si appiatisce, perdendo le
peculiarità dei primi anni di vita; tuttavia,
lidentificazione delle caratteristiche faciali in età
adulta risulta fondamentale per individuare casi isolati della
malattia, spesso seguiti per un difetto cardiaco congenito
apparentemente isolato, che hanno però un rischio elevato di
avere figli affetti (il rischio di ricorrenza di cardiopatia
congenita è del 50%). Il riconoscimento della facies è
poi fondamentale per sottoporre questi soggetti a un counseling
genetico6.
Caratteristiche
meno frequenti sono poi anomalie renali e vascolari (emorragie
cerebrali), endocrinopatie, ritardo mentale7-8, e anomalie
auricolari (alterazioni delle ossa temporali, otite media cronica,
sordità, displasia dei canali semicircolari posteriori)11.
La
gestione di questi pazienti dipende dalla severità
delle manifestazioni; di fondamentale importanza è un supporto
nutrizionale adeguato, il quale previene il progressivo
deterioramento della funzionalità epatica, aumentando la quota
energetica necessaria alle sue funzioni di sintesi, deposito e
detossificazione12. Questo comprende una dieta ipolipidica
con supplementazione di vitamine liposolubili, acidi grassi
essenziali e trigliceridi a catena media. La terapia standard per
il prurito include luso di antistaminici, resine leganti
gli acidi biliari, rifampicina, fenobarbital e acido
ursodesossicolico (molto efficace anche nella cura degli xantomi),
utilizzati singolarmente o in varie combinazioni. Nelle forme che non
rispondono a nessun tipo di terapia farmacologica, con funzionalità
epatica ancora buona, esiste la possibilità di effettuare un
intervento di diversione biliare parziale esterna. In questa malattia
è presente unalterazione della secrezione della bile, a
causa della paucità dei dotti biliari, per cui i suoi
costituenti refluiscono nel plasma; tale intervento permette uno
shunt della bile, impedendo il deterioramento della funzionalità
epatica, e permettendo una riduzione importante degli acidi biliari
circolanti, con conseguente miglioramento del prurito. La diversione
biliare consente inoltre una riduzione del colesterolo totale
mediante un meccanismo simile a quello delle resine anioniche
(colestiramina)9.
Per
quanto riguarda i difetti cardiaci, è molto spesso necessaria
una correzione cardiochirurgica. La prognosi dipende dalla
severità del coinvolgimento cardiaco ed epatico, oltre che da
eventuali infezioni, con una mortalità media del 17-30%. Nei
pazienti refrattari ad ogni trattamento medico cè
lindicazione al trapianto di fegato; la sopravvivenza a 20
anni è del 75%, mentre cala al 50% in quelli non trapiantati.
I pazienti con sindrome di Alagille rappresentano il 6% circa di
tutti i trapianti di fegato in età pediatrica 7-8.
Fegato
Cuore
Occhi
Scheletro
Facies |
Colestasi,
ittero, prurito
Insufficienza
epatica
Stenosi
periferica a. polmonare
Embriotoxon
posteriore
Emivertebre/fusioni
vertebrali
Anomalia/agenesia
ulnare
Ipoplasia
delle falangi distali delle dita
Prominenza
bozze frontali
Ipertelorismo |
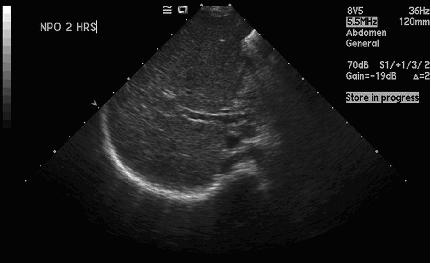
Figura
2. Ecografia epatica dopo 2 ore di digiuno: assente dilatazione
vie biliari intraepatiche e mancata evidenziazione di dotto biliare
comune e colecisti
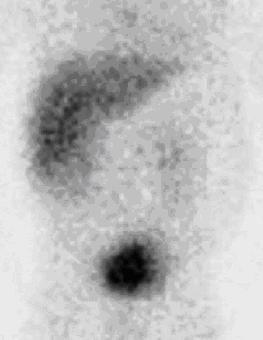
Figura
3. Colescintigrafia con Tecnezio: lieve ritenzione del
radionuclide a livello epatico, senza escrezione nelle vie biliari,
colecisti o intestino; esso viene evidentemente eliminato per via
renale, come dimostra laccumulo del tracciante in vescica.

Bibliografia
1.Sanderson
E, Newman V, Haigh SF, Baker A, Sidhu PS, Vertebral anomalies in
children with Alagille syndrome: an analysis of 50 consecutive
patients, Pediatr Radiol 2002;32:114-119.
2.Delgado
A, Mokri B, Miller GM, Butterfly vertebrae, J Neuroimaging
1996;6:56-58.
3. Krantz
ID, Piccoli DA, Spinner NB, Alagille syndrome, J Med
Genet1997;34:152-157.
4. Ryan
RS, Myckatyn SO, Reid GD, Munk P, Alagille syndrome: case-report with
bilateral radio-ulnar synostosis and a literature review, Skeletal
Radiol 2003; 32:489-491.
5. Ghidini
A, Incerti M, Andreani M, Alagille syndrome: prenatal sonographic
findings, J Clin Ultrasound 2007; 35:156-158.
6. Kamath
BM, Loomes KM, Oakey RJ, Emerick EM, Conversano T, Spinner NB,
Piccoli DA, Krantz ID, Facial features in Alagille syndrome:
specific or cholestasis facies?, American Journal of Medical Genetics
2002; 112:163-170.
7. Sze
DY, Esquivel CO, SIR 2008 annual meeting film panel case: Alagille
syndrome, J Vasc Interv Radiol 2008; 19:1278-1281.
8. Schwartz
R, Rehder K, Parsons DJ, Morrell DS, Intense pruritus and failure to
thrive in Alagille syndrome, J Am Acad Dermatol 2008;58:S9-S11.
9. Emerick
KM, Whitington PF, Partial external biliary diversion for intractable
pruritus and xanthomas in Alagille syndrome, Hepatology 2002; vol.35,
No 6:1501-1506.
10. Narula
P, Gifford J, Steggall MA, Lloyd C, Van Mourik IDM, Mckiernan PJ,
Willshaw HE, Kelly D, Visual loss and idiopathic intracranial
hypertension in children with Alagille syndrome, J Pediatr
Gastroenterol Nutr 2006; 43:348-352.
11. Koch
B, Goold A, Egelhoof J, Benton C, Partial absence of the posterior
semicircular canal in Alagille syndrome: CT findings, Pediatr Radiol
2006;36:977-979.
12. Rovner
AJ, Stallings VA, Piccoli DA, Mulberg AE, Zemel BS, Resting energy
expenditure is not increased in prepubertal children with Alagille
syndrome, J Pediatr 2006;148(5):680-682.
Vuoi citare questo contributo?