Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Novembre 2011 - Volume XIV - numero 9
M&B Pagine Elettroniche
Casi indimenticabili
Pelle
di leopardo
1Scuola
di Specializzazione in Pediatria, IRCCS Pediatrico Burlo Garofolo,
Università di Trieste
2Scuola
di Specializzazione in Genetica Medica, Università di Padova
|
Luca
viene alla nostra attenzione all'età di 16 anni. La sua storia
clinica si caratterizza per uno sviluppo psicomotorio nella norma
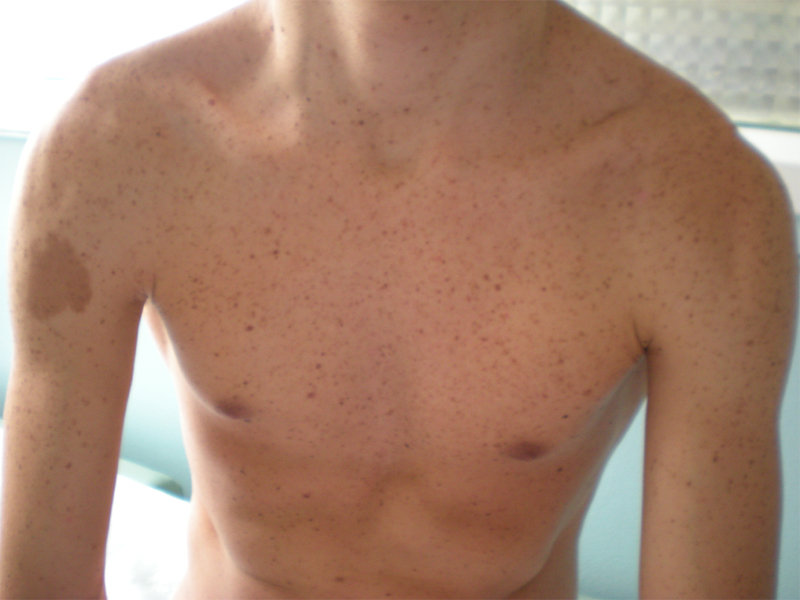
e per la comparsa di macchie caffè-latte a un anno di vita (9
lesioni > 0,5 cm) che inducono i medici ad avanzare l'ipotesi
diagnostica di neurofibromatosi di tipo 1 (NF1). A due anni di
vita subisce un intervento correttivo per la presenza di
criptorchidismo bilaterale. Successivamente si realizza la
comparsa di lentigginosi diffusa sulla cute di tutto il corpo,
con incremento del numero delle lesioni nel tempo e loro
diffusione anche in regione ascellare e inguinale. Non si
evidenziano noduli di Lisch alla visita oculistica. La crescita
staturo-ponderale si è sempre mantenuta al di sotto del 3°
percentile, in presenza di un'età ossea compatibile con l'età
cronologica. Altri reperti che emergono all'esame obiettivo
(EO) sono il prognatismo, il torace carenato e le scapole alate.
Al tracciato ECG si evidenzia un blocco di branca sinistro (in un
unico rilievo). Buono il rendimento scolastico.
Alcune
delle caratteristiche fenotipiche del ragazzo sono presenti anche
in sua mamma, la quale mostra all'EO la presenza di
lentigginosi diffusa e di 4 chiazze caffè-latte di diametro
superiore ai 15 mm; risulta negativa alla ricerca di noduli di
Lisch alla visita oculistica. Gli approfondimenti svolti sul
versante cardiologico rilevano anche un aneurisma aortico di
modesta entità e una lieve disfunzione mitralica. Nel complesso
nella mamma di Andrea non vengono soddisfatti i criteri minimi
per porre diagnosi di NF1.
Man
mano che Luca cresce e si sottopone ai
controlli periodici previsti per il follow-up della sua
condizione, il sospetto diagnostico di NF1 appare meno fondato.
Infatti, quando viene rivalutato clinicamente, quelle macchie,
che nei primi anni di vita erano state inquadrate come macchie
caffè-latte, non sono mai aumentate di numero e hanno assunto
nel tempo un aspetto frastagliato e irregolare. Anche le
lentiggini non hanno la topografia propria del frackilng
ascellare e inguinale ma sono molto più numerose in altri sedi
cutanee. Permane poi, l'assenza dei noduli di Lisch: nonostante
l'età di Luca sia oramai superiore ai 10 anni non c'è
traccia di noduli cutanei e l'anamnesi familiare è tutto
sommato muta. Nonostante l'espressività possa essere molto
variabile, a questo punto i conti proprio non tornano
L'ipotesi diagnostica più verosimile è allora quella della
lentigginosi familiare e viene pertanto avviata l'indagine
molecolare inerente, che rivela presenza di una mutazione
patogenetica del gene PTPN11, responsabile della sindrome
di Leopard!
La
sindrome di LEOPARD è una
patologia a trasmissione AD, il cui nome è l'acronimo inglese
delle caratteristiche della sindrome: Lentigines,
ECG conduction abnormalities, Ocular hypertelorism, Pulmonic
stenosis, Abnormal genitalia, Retardation of growth and
sensorineural Deafness. Spesso si
osservano anche un ritardo nell'accrescimento staturo-ponderale
(< 3° percentile), macchie caffè-latte, criptorchidismo,
prognatismo, petto carenato e scapole alate. L'espressività è
variabile, la penetranza completa e la prognosi ben più
favorevole che nella NF1.
E'
stato indimenticabile perché si è avuto il buonsenso di mettere
in discussione una diagnosi già posta, mostrando l'importanza
della rivalutazione clinica di un paziente anche in corso di
follow-up, qualora una diagnosi si faccia meno convincente.
Inoltre dimostra l'importanza dell'esame obiettivo di reperti
facilmente accessibili come le lesioni iperpigmentate
(dimensione, numero, distribuzione, aspetto dei margini, epoca
della comparsa, evoluzione nel tempo) e della loro diagnosi
differenziale, anche alla luce di altri segni e sintomi di
accompagnamento. E nonostante questo, talvolta, la clinica da
sola non basta per arrivare a una diagnosi e il test genetico si
rivela cruciale per poi potersi esprimere correttamente in
termini di prognosi.
Bibliografia
di riferimento
|
 |
Vuoi citare questo contributo?