Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Settembre 1999 - Volume II - numero 7
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Sindrome
di Teresa Kindler
Divisione
di Pediatria Ospedale di Bentivoglio ASL Bologna Nord
*Servizio
di Neuropsichiatria Infantile ASL Bologna Nord
Teresa
Kindler's syndrome
The
Case
SBH is a
Tunisian child, born from I degree cousins (negative familiar
history). She was referred to us when she was 14 years old with a
diagnosis of Goldscheider's disease.
At the
objective examination she showed: poikiloderma with skin atrophy
mainly to the members, where microtraumas had produced small bullous
lesions; gingivostomatitis; signs of dermatitis due to face
photosensitivity; low stature (130 cm); delayed puberty (II degree of
Tanner); drumstick-shaped hands; mild mental deficit; significant
knee valgoid condition, especially on the left side; dactylion to the
3rd-4th fingers of the foot; hyperglycinuria (44% of total
aminoaciduria).
The
Problem
At birth,
a differential diagnosis between Goldscheider's disease and T.
Kandler's syndrome is difficult because of the presence of bullous
lesions in both pictures; afterwards, however, the lesions due to
Kandler's disease tend to disappear, and they leave a specific
sensitivity to microtraumas which, together with photophobia, stature
deficit and metal deficit, can lead to differential diagnosis.
The
Contribution
Ultramicroscopic
study, assessment of hyperglycinuria (not described so far),
recessive autosomic familiarity, very rich clinical picture,
contribution to the diagnosis of Goldscheider's disease.
Articolo
Introduzione
La
sindrome di Teresa Kindler, descritta per la prima volta nel 1954 da
Teresa Kindler (1), è caratterizzata da poichilodermia
generalizzata e progressiva con atrofia cutanea, presenza di
vescicole cutanee alle estremità e fotosensibilità.
Questa forma può essere differenziata dalla epidermolisi
bollosa congenita e dalle altre poichilodermie bollose ereditarie;
oltre alle alterazioni cutanee sono presenti ritardo mentale e di
crescita e iperaminoaciduria generalizzata.
Sin dalle
prime descrizioni sono stati riportati pochissimi casi al mondo e gli
studi ultrastrutturali sono stati eseguiti solo su una decina di
pazienti (7).
Questa
sindrome può essere differenziata dalla epidermolisi bollosa
congenita, ma solo in età tardiva, in quanto alla nascita
presenta un quadro molto simile a quello della forma bollosa. Dopo i
primi anni di vita la tendenza a formare bolle diminuisce ed esse
compaiono sulle superfici distali in seguito a sfregamenti o piccole
lesioni. Successivamente, la pelle delle mani, dei polsi, dei piedi,
delle caviglie, dei gomiti e delle ginocchia mostra atrofia
superficiale e zone sparse pigmentate. Le zone di atrofia sono molto
più pronunciate alle estremità, con aspetto a "carta
di sigaretta" soprattutto nella zona dorsale dei piedi e delle
mani.
Nella
maggior parte dei casi descritti, i genitori erano consanguinei, il
che indica una ereditarietà recessiva (4,6). Sono stati
descritti anche casi a trasmissione autosomica dominante.
Il
caso
LS.B.H. è
giunta alla nostra osservazione all'età di 14 anni, appena
immigrata dalla Tunisia, con diagnosi di epidermolisi bollosa
congenita recessiva tipo Hallopeau-Siemens, effettuata nel paese di
origine.
I
genitori sono cugini di 1° grado .
All'esame
obiettivo era possibile evidenziare, più che la presenza di
bolle, una poichilodermia con atrofia cutanea prevalentemente a
carico delle estremità(fig.1).
fig.1
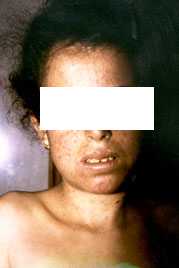
A queste
importanti lesioni si associano:
importante
difetto di crescita (altezza cm 130,5; peso kg 21) associata a
ritardo puberale (II stadio di Tanner) (fig.2);
fig.2
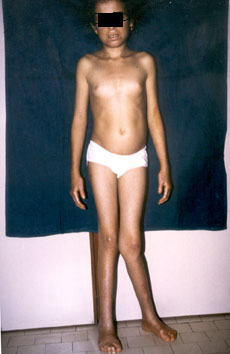
una
importante gengivostomatite;
segni di
pregresse alterazioni da fotosensibilità al volto;
valgismo
di ginocchio particolarmente importante a sinistra (fig. 2);
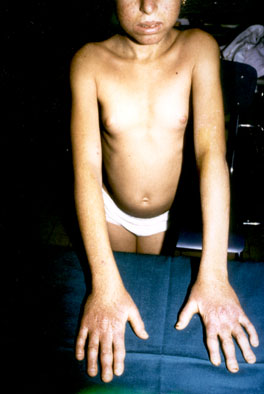
dita a
bacchette di tamburo (fig.3-4);
fig.3
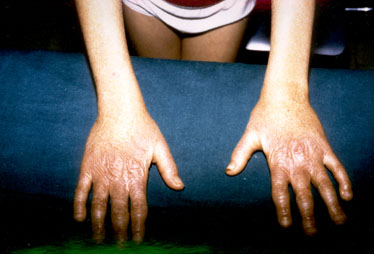
fig.4
sindattilia
del III e IV dito di entrambi i piedi;all'esame radiologico: lieve
displasia bilaterale dell'anca; età ossea: 8 anni.
Abbiamo
praticato gli accertamenti ematochimici routinari, cariotipo, EMA ed
AGA, ricerche autoimmunitarie, infettivologiche (sifilide, ep.C,
ep.B, tampone del cavo orale), ormonali (tiroide e paratormone), IgE
ed il dosaggio degli aminoacidi urinari; tutti gli esami sono
risultati essere nella norma, ad eccezione di una elevata
concentrazione di glicina urinaria, che rappresentava da sola il 44%
dell'aminoaciduria totale.
Gli
accertamenti radiologici mostravano invece una lieve displasia
bilaterale delle anche, ossa delle dita delle mani a bacchetta di
tamburo, valgismo bilaterale delle ginocchia e una età
scheletrica approssimativamente compatibile con 8 anni di età
anagrafica.
La
consulenza dermatologica, associata a biopsia cutanea per lo studio
ultrastrutturale, ha permesso di diagnosticare una sindrome di
Teresa-Kindler associata in questo caso a ritardo di crescita,
ritardo puberale, grave dismorfismo di tutti gli arti ed in
particolare degli inferiori con valgismo soprattutto a carico del
ginocchio sinistro che, da valutazione ortopedica, sarà
necessario correggere chirurgicamente.
Associato
a ciò, durante la degenza è emersa la presenza di un
ritardo cognitivo di difficile inquadramento per le ovvie difficoltà
di comunicazione.
Il
deficit intellettivo, già osservato in letteratura, risultava
essere ascrivibile ad un ritardo mentale di grado lieve, secondo
quanto descritto dall'ICD10, rispetto all'età ossea. Per tale
accertamento è stata utilizzata una metodica applicabile a
bambini affetti da difficoltà di comunicazione verbale,
utilizzando unicamente prove di performance della WISC-R, affinché
la difficoltà della lingua straniera non inficiassero
l'obiettività della valutazione NPI.
Attualmente
sono in corso accertamenti genetici tra la famiglia di S.B.H e quella
di una bambina della nostra provincia, nata anch'essa da genitori
consanguinei, affetta da sindrome di Teresa Kindler, allo scopo di
confrontare il materiale genetico cromosomiale.
Istologia
Gli studi
istologici ed ultra strutturali mostravano la separazione sub basale
della lamina ed in particolare il tetto della vescicola è
costituito dalla lamina basale e da materiale fibrillare amorfo
connessi. Una scoperta inusuale è la presenza di fibroblasti
nella regione subpendimale con caratteristica di attivazione, con
apparenza di turgidità, con cisterne del reticolo endoplasmico
rugoso allargate ed aumento nel contenuto dei ribosomi liberi e
lisosomi che sono strettamente incollati ai cheratinociti basali.
La
giunzione dermo-epidermica è lievemente interessata e mostra
fissurazioni focali della lamina lucida e reduplicazione ed
interruzione della lamina densa.
Il
contributo
Il nostro
caso si aggiunge alla esigua casistica mondiale. Conferma la
possibilità di distinguere (per la presenza di
fotosensibilità, di interessamento mucosale e per la
formazione di bolle post-traumatiche, la poichilodermia di Teresa
Kindler dalla epidermolisi bollosa ereditaria. Conferma la
trasmissione di tale patologia come carattere autosomico in genitori
consanguinei con albero genealogico indenne, dunque, verisimilmente,
in via recessiva. Lo studio istologico ultrastrutturale delle lesioni
cutaneee contribuisce alla diagnosi differenziale, colloca la lesione
fondamentale subito al di sotto della lamina basale, con una
componente fibroblastica associata sui generis. Il nostro caso
aggiunge alla sindrome descritta dalla letteratura l'osservazione di
alterazioni scheletriche complesse e di una iperglicinuria.
Bibliografia
1)
Kindler T: Congenital poikilodermia with traumatic bulla formation
and progressive cutaneous atrophy. Br. J. Dermatol 66,
104-111, 1954.
2)
Hovnanian A, Blanchet-Bardon C, de Prost Y: Poikilodermia of Theresa
Kindler : Report a case with ultrastructural study , and review of
the literature. Pediatr Dermatol 6, 82-90, 1989
3) Forman
AB , Prendiville JS, Esterly NB et al: Kindler syndrome: Report of
two cases and review of the literature. Pediatr. Dermatol. 6,
91-101, 1989
4)
Passwell J, Zipperkowski L , Katznelson D et al: A syndrome
characterized by congenital ichthiosis with atrophy, mental
retardation, dwarfism, and generalized aminoaciduria. J
Pediatr 82(3), 466-471, 1973
5) Bordas
X, Palou J, Capdevilla JM, Mascaro JM: Kindler's syndrome. J Am
Acad Dermatol 6, 263-265, 1982
6) Chong
HT: Ichthyosiform erytroderma,hair shaft abnormalities, and mental
and growth retardation. Arch Derm 104, 4-13, 1971
7)
Patrizi A, Pauluzzi P, Neri I, Trevisan G, Badiali De Giorgi L,
Pasquinelli G: Kindler Syndrome: report of a case with
ultrastructural study and review of a literature. Pediatr
Dermatol 13(5), 397-402, 1996
Vuoi citare questo contributo?