Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Aprile 2010 - Volume XIII - numero 4
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Una
strana crisi isterica
1UOC
di Pediatria, Ospedale S. Camillo de Lellis, Rieti
2Facoltà
di Medicina e Chirurgia, Università La Sapienza, Roma
In una
sera di febbraio giunge in reparto, su richiesta del pediatra
curante, un bambino di 9 anni e mezzo, affetto da dolori addominali
insorti da circa 48 ore e associati a numerosi episodi di vomito
alimentare; non febbre né alterazioni dellalvo.
Lanamnesi
familiare è negativa, ha una sorella di 11 anni in apparente
buona salute.
Lanamnesi
personale è la seguente: nato a termine, parto eutocico, peso
alla nascita 2800 g, allattamento materno per 5 mesi, latte vaccino
poi. Divezzamento a 4-5 mesi, sviluppo psicomotorio nella norma; un
ricovero per enterite acuta circa quattro anni prima e indagini per
sospetta mononucleosi a causa di un transitorio aumento delle
transaminasi (sospetto poi non confermato). Il peso attuale è
di 23,5 kg (<10 °C).
Durante
la notte persiste vomito per cui, il mattino seguente il ricovero,
esegue esami di routine, risultati nella norma, tranne amilasi
lievemente aumentata (237 con valori normali fino a 220); inizia
infusione di glucosata alternata a bilanciata. Le condizioni generali
sembrano buone; il bambino è collaborante, vigile, non ha
febbre e, allesame obiettivo, non presenta segni neurologici;
la palpazione addominale rileva modico dolore in sede periombelicale.
Non siamo preoccupati: ci sembra una delle tante gastriti o
gastroenteriti virali che in questo periodo giungono in consulenza o
in ricovero. In tarda mattinata il bambino presenta però una
crisi di agitazione psicomotoria con importante aggressività
rivolta alla madre (ci sono ciocche di capelli materni a terra!); la
signora dice che il figlio è insofferente e ribelle a causa
della flebo e che anche a casa se la riprende spesso con lei, dato
che vivono in ambiente rurale isolato e gli amici non sono vicini;
dopo la crisi presenta sonno con volto atteggiato al sorriso. Le
spiegazioni materne non ci convincono del tutto, non ci è mai
capitata una cosa simile; non disponiamo di un consulente di
neuropsichiatria infantile allinterno dellospedale,
consultiamo perciò uno psichiatra delladulto che
interpreta laccaduto come possibile crisi isterica. Viene
praticata una fiala di benzodiazepina. Una consulenza oculistica per
fundus oculi risulta negativa. Più tardi il bambino si
sveglia, va autonomamente in bagno, alla visita delle ore 17 è
sul letto, in posizione genu pettorale, si rifiuta di parlare con noi
e non vuole farsi visitare. Si effettua un breve consulto di reparto:
chi ha maggiore esperienza non propende per unencefalite
infettiva (3 di VES e 0,1 di PCR); anche una sindrome di Reye non
sembra ipotizzabile (transaminasi nella norma); tossici vengono
negati con decisione; qualcuno di noi è sicuro che si tratti
solo di isteria e propone benzodiazepine per os per le ore
successive. Si soprassiede alla puntura lombare, anche perché
nel frattempo giungono in ricovero due emergenze: una polmonite in
glomerulonefrite acuta e una meningite.
A fine
turno, si sono fatte le ore 20 ormai, il medico torna a controllare
quello strano caso clinico e trova il piccolo obnubilato, ha perduto
urine, presenta ipertono e riflessi OT accentuati. La madre riferisce
che prima di addormentarsi il piccolo ha ancora
presentato due crisi di agitazione. Viene eseguita una TAC - encefalo
che risulta negativa e vengono ripetuti in urgenza esami
ematochimici: transaminasi, creatinina, elettroliti sono nella norma,
la glicemia è 142 mg%, la bilirubina è aumentata
modicamente (2,11 con 0,7 di diretta). Lammoniemia, pur
richiesta, non ci perviene (ahimè) per inconveniente tecnico e
il laboratorio ci chiede un secondo campione. Intanto il paziente è
preso in carico dai colleghi della rianimazione: è in coma
vero, presenta midriasi con riflessi pupillari torpidi, congiuntivo
corneali presenti, faringo-laringei presenti, osteotendinei
accentuati, Babinski assente, respiro valido, sensibilità
dolorifica assente. LECG rileva aritmia sinusale respiratoria.
Si è avviato il trasferimento presso centro di terzo livello,
si pratica bicarbonato sulla scorta dellemogas e si
soprassiede al reinvio di sangue per lammoniemia: il centro
mobile attende e il piccolo deve essere intubato per il trasporto. In
tarda serata affidiamo il bambino alla TIP che ci ha dato
disponibilità di posto letto: la puntura lombare, le ricerche
virologiche e tossicologiche risultano negative, ma la TAC encefalo
ripetuta lindomani mattina dimostra imponente edema cerebrale,
lEEG è piatto. Ingannati dalla normalità delle
transaminasi (contro la sindrome di Reye) e indirizzati verso
lipotesi virale dallincremento dellamilasi, i
colleghi della TIP eseguono lammoniemia solo in seconda
giornata: è alle stelle! Partono così le indagini
metaboliche, in vivo e in vitro: la diagnosi finale sarà di
intolleranza alle proteine con lisinuria. Certamente letà
avanzata del paziente non ha reso facile il giusto
sospetto. Col senno del poi si interpretano nella giusta luce gli
episodi di irrequietezza che risultano dalla storia remota (peraltro
minimizzati dalla madre) e linsufficienza ponderale; lassenza
di altri sintomi anamnestici o fisici non ha purtroppo aiutato.
Lintolleranza
alle proteine con lisinuria è una malattia autosomica
recessiva, dovuta a un difetto di trasporto degli aminoacidi dibasici
(lisina, arginina, ornitina) attraverso la membrana basolaterale
delle cellule epiteliali dellintestino e del rene. SLC7A7 è
il gene responsabile: codifica per la catena leggera del
trasportatore y+ degli aminoacidi cationici1. La malattia è
stata descritta per la prima volta in Finlandia, dove è
riportata una frequenza di un caso su 60.000 nati; due altre regioni
geografiche con prevalenza relativamente alta sono il Sud Italia e il
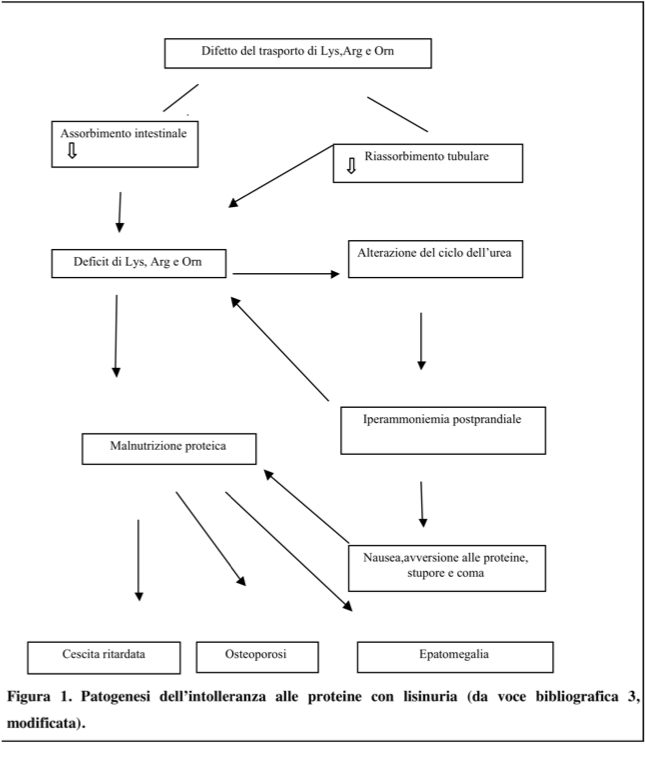
Giappone. Conseguenze del difettoso trasporto di arginina, lisina e
ornitina sono il mancato assorbimento intestinale e la perdita
urinaria di questi aminoacidi; la carenza di arginina e ornitina è
responsabile di un blocco funzionale del ciclo dellurea
(principale via di escrezione dellazoto nelluomo), con
accumulo di ioni ammonio nei fluidi biologici, come illustrato in
Figura 1; alla scarsa disponibilità di
lisina si attribuisce invece losteoporosi, reperto costante
dopo i primi anni di vita2.
Classicamente
i pazienti affetti sono asintomatici durante lallattamento al
seno, dato il basso introito proteico con il latte materno; subito
dopo lo svezzamento compaiono vomito e diarrea. Lappetito è
scarso, la crescita stentata e, se si forza lassunzione di
maggiori quantità di proteine, si manifestano i segni clinici
dovuti alliperammoniemia, fino a crisi di letargia e coma.
Come meccanismo protettivo, dopo la prima infanzia i piccoli
tipicamente sviluppano forte avversione per i cibi ricchi di
proteine, con una possibile e temporanea regressione dei sintomi che
può far sfuggire la diagnosi nella prima e seconda decade di
vita; allesame obbiettivo i reperti di più frequente
osservazione sono lepatosplenomegalia, lipotonia e
lipotrofia muscolare. Lo sviluppo mentale è normale
nella maggior parte dei casi salvo che eventi iperammoniemici acuti
non esitino in danni neurologici3. Uno studio
multicentrico italiano ha esaminato 23 pazienti affetti da IPL: ne è
risultata una marcata variabilità fenotipica, con possibile
interessamento multisistemico accanto ai segni tipici, come
illustrato in Tabella I.
Segni
tipici
Avversione
alle proteine 11/23
Visceromegalia
19/23
Crisi
neurologiche 6/23
Deficit
di crescita 10/23
Segni
atipici
Interessamento
polmonare 7/15
Interessamento
renale 8/15
Alterazioni
midollari 8/10
Dolori
addominali pancreatite 3/17
Tratto
da voce bibliografica 3, modificata. |
Tabella
1. Intolleranza alle proteine con lisinuria: segni tipici e
atipici alla diagnosi: casistica italiana
Attualmente
si ritiene che proprio le complicanze elencate fra i segni atipici
possano rendere meno benigna di quanto atteso la malattia in
trattamento, compromettendone la prognosi: infatti esse, sia pur
rare, non sono sensibili alla terapia dietetica e il loro meccanismo
patogenetico è ancora oscuro. La più severa è
una pneumopatia interstiziale che si presenta con progressiva
dispnea, tosse, cianosi, opacità interstiziali alla
radiografia del torace e proteinosi alveolare allanatomia
patologica3. La proteinosi alveolare ha presentato una
ricorrenza infausta in un paziente con IPL sottoposto a trapianto
cuore-polmoni4. Altre possibili complicanze sono pancreatite,
interessamento renale (da una lieve proteinuria a una grave
nefropatia con alterazioni del glomerulo e del tubulo),
autoeritrofagocitosi midollare, anomalie del sistema immunitario
(presenza di cellule LE, anticorpi anti-DNA, ipergammaglobulinemia o
basse concentrazioni seriche di immunoglobuline, alterazioni
linfocitarie).
Dallesame
delle diagnosi iniziali nei pazienti della casistica sopra citata
emerge che in alcuni casi i sintomi e i dati laboratoristici sono
stati fuorvianti: lipotonia muscolare e laumento di
LDH-CK hanno posto il sospetto di malattia mitocondriale, la
visceromegalia, linterstiziopatia e leritroblastofagocitosi
midollare hanno indirizzato verso malattie da accumulo,
linsufficienza accrescitiva, la diarrea cronica e
lipoalbuminemia hanno suggerito il morbo celiaco; quanto detto
è riassunto in Tabella 2.
Malattia
di Gaucher 3
Malattia
di Niemann-Pick tipo B1
Mucopolisaccaridosi
atipica 2
Sospetta
malattia da accumulo 1
Glicogenosi
1
Sospetta
malattia mitocondriale 1
Sospetta
patologia ematologia 1
Malattia
celiaca 1
Tratto
da voce bibliografica 3, modificata. |
Liperammoniemia
dellIPL deve esser differenziata da quella legata a difetti
enzimatici ereditari del ciclo dellurea, da quella secondaria
a insufficienze epatiche di varia origine o ad altri errori congeniti
del metabolismo, quali le acidurie organiche e i difetti di
ossidazione degli acidi grassi.
-
lammoniemia è normale a digiuno, ma aumenta dopo pasti
ricchi di proteine, tanto più quanto maggiore è
lintroito proteico;
- le
concentrazioni plasmatiche di lisina, arginina e ornitina sono basse
(o normali-basse);
-
lescrezione urinaria e la clearance degli aminoacidi cationici
(specialmente lisina) sono aumentate;
- le
concentrazioni di serina, glicina, prolina, citrullina, alanina e
glutamina sono sempre aumentate;
- LDH,
ferritina serica e TBG sono usualmente aumentati;
- reperti
aggiuntivi aspecifici possono esser trovati: trombocitopenia, lieve
anemia, leucopenia.
-
sospensione dellapporto proteico, mantenendo però un
adeguato apporto calorico per evitare il catabolismo endogeno;
-
somministrazione ev di arginina cloridrato e di farmaci quali
benzoato e fenil-butirrato che aumentano lescrezione
dellazoto attraverso lattivazione di vie alternative.
Se non
cè risposta entro le prime ore a queste iniziali misure
terapeutiche, è necessario ricorrere a tecniche di depurazione
extracorporea onde evitare sequele permanenti; lammonio
esplica azione tossica sul SNC attraverso meccanismi che coinvolgono
la sintesi dei neurotrasmettitori e il metabolismo energetico
cellulare5.
- dieta
ipoproteica: va impostata a seconda delletà del
paziente e attentamente individualizzata; nel primo anno 1-1,5
g/kg/die, durante linfanzia 1 g/kg/die, nelladulto
0,5-0,7 g/kg/die. È raccomandato, nel primo biennio di vita,di
limitare strettamente lapporto proteico per evitare
innalzamenti dellammoniemia mantenendo sotto stretto controllo
la curva di crescita. Successivamente, fino alla pubertà, va
calcolata la tolleranza massima individuale allazoto; nel
periodo puberale (rischio di ricomparsa di instabilità
metabolica) lapporto di proteine va nuovamente limitato;
-
supplementazione con citrullina (2,5-8,5 g/die) cercando la
più bassa dose efficace in funzione della risposta clinica e
biochimica:lescrezione urinaria di acido orotico sembra un
sensibile strumento per aggiustare il trattamento. Il razionale della
citrullina è nel fatto che è assorbita normalmente e
metabolizzata ad arginina e ornitina; il livello plasmatico
dellarginina deve mantenersi fra 80 e150 µmol/l;
-
benzoato di sodio (100-250 mg/kg/die per os) e fenilbutirrato
agli stessi dosaggi sono utilizzabili da soli o in combinazione. Non
hanno tossicità ma effetti indesiderati o palatabilità
ne limitano spesso limpiego.
La dieta
deve comunque essere caloricamente adeguata per prevenire condizioni
di catabolismo. I pazienti vanno monitorizzati per i parametri
auxologici, la maturazione ossea, lo sviluppo intellettivo; tecniche
di imaging possono integrare la valutazione clinica. Vanno in
parallelo monitorizzati gli indici biochimici e nutrizionali:
ammoniemia e aminoacidi su profilo diurno, transaminasi,
colinesterasi, bilirubina, albumina, sodio calcio, fosfatasi alcalina
e acido orotico, LDH, ferritina. Vanno individuate e prontamente
trattate le condizioni carenziali (vitamine,oligoelementi); un buon
indicatore dellapporto di aminoacidi essenziali è la
treonina, il cui livello deve essere superiore a 80 µmol/l5.
Per le
complicanze sistemiche probabilmente sostenute da meccanismi
autoimmuni sono state proposte sia la terapia cortisonica che quella
con immunoglobuline ev6.
Identificazione
dei portatori sani e diagnosi prenatale nelle gravidanze a rischio
Lidentificazione
dei portatori sani e la diagnosi prenatale nelle gravidanze a rischio
sono rese possibili da indagini di genetica molecolare, che in Italia
sono state effettuate dal gruppo di Napoli (G. Sebastio, M.P.
Sperandeo, G. Parenti, G. Andria), Università Federico II.
Messaggi
chiave
|
Bibliografia
1.
Borsani G, Bassi MT, Sperandeo MP, et al.
SLC7A7, encoding a putative permease related protein is mutated in
patients with lysinuric protein intolerance. Nat
Genet 1999;21:297-301.
2.
Parto K, Penttinen R, Paronen I, Pelliniemi L, Simell O. Osteoporosis
in lysinuric protein intolerance. J Inherit
Metab Dis 1993;16:441-50.
3. Scala
I, Parenti G, Sebastio G, Andria G. Intolleranza alle proteine con
lisinuria: una malattia multisistemica curabile, ma non sempre.
Prospettive in pediatria 2001;31:133-41.
4.
Santamaria F, Brancaccio G, Parenti G, et al.
Recurrent fatal pulmonary alveolar proteinosis after heart-lung
transplantation in a child with LPI. J Pediatr
2004;145:268-72.
5.
www.ospedalebambinogesu.it. Portale 2008 Malattie rare Iperammoniemie
ereditarie.
6.
Dionisi Vici C, De Felice L, El Hachem M, et al. Intravenous immune
globulin in LPI J Inher Metab Dis 1998;21:95-102.
Vuoi citare questo contributo?