Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Febbraio 2004 - Volume VII - numero 2
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Epilessia
benigna occipitale a esordio precoce
Cattedra
di Pediatria - DPMSC -Policlinico Universitario di Udine
Key
Words: occipital seizure, Clinical manifestation, prognosis
Three
cases of early-onset benign occipital epilepsy (BOE) are reported in
two three year-old female children and in one ten year-old female
children and in one ten year old boy. Early onset BOE is among the
most frequent epilepsies in childood. Its typical features are the
long duration of the first episode, which usually starts in a normal
child during sleep or on waking up and frequently remains the only
one. Inter-critical EEG patterns are variable and vary over time. The
course is benign, usually self-limited within primary school-age
L'epilessia
benigna occipitale precoce rappresenta un quadro di epilessia focale
idiopatica dell'infanzia non raro, che si caratterizza per aspetti
clinici ed elettroencefalografici peculiari e per l'evoluzione
benigna. Il suo riconoscimento da parte del pediatra sembra opportuno
e pertanto presentiamo la descrizione di tre fra i casi giunti a
nostra osservazione, che risultano particolarmente esemplificativi.
M.E.
Bambina con anamnesi familiare e personale non significativa. All'età
di tre anni, durante il riposo pomeridiano, compaiono tosse, conati
seguiti da un vomito, deviazione tonica dello sguardo a sinistra,
fluttuazione dello stato di coscienza con graduale perdita di
contatto senza manifestazioni cloniche. Viene portata al nostro
reparto dove giunge dopo mezz'ora dall'insorgenza dei sintomi: si
presenta afebbrile, priva di coscienza, mantiene deviazione dello
sguardo tonica a sinistra ma senza altri segni di lato, le condizioni
generali sono buone. L'EEG dimostra uno stato epilettico con
partenza focale destra che si risolve con somministrazione di
diazepam EV.
Al
risveglio la bambina è perfettamente normale come obiettività
e comportamento, che risulta adeguato all'età. La TC è
negativa. Negative le sierologie per EBV, HZV, Coxsackie. Vaccinata
per MMR.
L'EEG a
distanza di qualche giorno evidenzia attività lenta e ampia
subcontinua in area parieto-occipitale destra e sporadiche onde
aguzze e punte trifasiche indipendenti centro-temporali
controlaterali.
Due mesi
dopo, sempre nel sonno pomeridiano, compare un episodio simile,
questa volta con deviazione tonica dello sguardo a destra e qualche
clonia brachio-faciale destra, durato circa mezz'ora e risolto nel
nostro reparto con la somministrazione di diazepam EV; residua
transitoriamente un'emiparesi faciale omolaterale postcritica.
Viene
eseguita una RMN cerebrale che risulta normale.
Nell'ipotesi
di una forma di epilessia focale benigna, tenendo conto della clinica
e degli aspetti EEG (fig.1:
anomalie lente occipitali sinistre coesistenti con sporadiche punte
lente centrotemporali controlaterali), si concorda con i genitori di
non avviare per il momento alcuna terapia: in effetti la bambina non
presenta più manifestazioni critiche e va incontro a uno
sviluppo perfettamente normale con ottimo profitto scolastico.
I
periodici controlli EEG documentano persistenza delle anomalie
segnalate con alterna prevalenza di lato e localizzazione (occipitale
o rolandica) fino a quando il tracciato risulta perfettamente
normalizzato all'età di 9 anni.
M.C.
Bambina di 3 anni e otto mesi, con storia familiare e personale non
significative, alla fine del riposino pomeridiano presso la scuola
materna viene richiamata ma non risponde e presenta deviazione del
capo e dello sguardo a destra, perdita di coscienza, successivamente
clonie generalizzate seguite da paresi destra che si risolvono in
ospedale dopo circa un'ora dall'insorgenza.
Al
risveglio l'obiettività generale e neurologica risultano
normali ed il comportamento adeguato all'età.
L'EEG
dimostra attività lenta delta in area parieto-occipitale
sinistra con presenza di punte lente parietali a sinistra.
Viene
consigliata l'osservazione clinica senza terapia e programmata una
RMN che risulta negativa.
Due anni
dopo presenta una breve crisi nel sonno con vomito e deviazione
oculare destra, per una decina di minuti. L'anno successivo si
manifesta ancora una crisi nel sonno notturno: vomita, si mette a
sedere con aspetto assorto, presenta fluttuazione della coscienza,
deviazione dello sguardo a sinistra, ancora vomito, ipotonia diffusa,
viene portata in ospedale dove la crisi viene risolta con diazepam.
Al risveglio la situazione è perfettamente normale.
La
bambina non presenta più crisi e lo sviluppo psicomotorio si
conferma normale.
L'EEG
presenta periodiche variazioni (fig.2:
si evidenziano onde lente aguzze subcontinue parieto-occipitali a
destra e triplette di punte-onda parieto-occipitali a sinistra) fino
a definitiva normalizzazione al controllo dei 9 anni.
C.S.
Ragazzino di 10 anni con storia personale non significativa e
familiarità per epilessia (zio paterno).
Poco dopo
il risveglio del mattino, viene trovato dai genitori accovacciato in
corridoio con capo e sguardo deviati a sinistra. Viene trasportato a
letto dal padre che descrive fluttuazioni della coscienza, iniziale
ipotonia e successivo aumento del tono, sempre con deviazione del
capo e dello sguardo a sinistra. Viene trasferito dell'equipe del
118 ad un ospedale periferico dove inizia a presentare scosse
tonico-cloniche all'emisoma di sinistra, nistagmo a sinistra,
morsus, perdita di urine. La somministrazione di diazepam EV risolve
solo parzialmente la crisi, per cui viene infuso tiopentale, il
ragazzo viene intubato ed inviato in terapia intensiva presso il
nostro ospedale, dove viene avviato valproato EV. Trasferito nel
nostro reparto non ha più avuto disturbi, nonostante la
graduale sospensione del valproato.
L'EEG
(fig.3)
ha dimostrato un'attività postcritica lenta, ampia e
continua sulle derivazioni occipitali con prevalenza a destra, che è
andata riducendosi, pur senza scomparire del tutto nei successivi
controlli, mentre nel sonno lento si documentavano punte rolandiche
all'emisfero controlaterale.
I
genitori hanno successivamente ricordato due precedenti episodi in
cui, sempre durante la notte, avevano trovato il figlio seduto sul
letto con sguardo fisso, ipotonico, non responsivo, ma si era poi
rapidamente riaddormentato. In ogni caso nel follow-up non si sono
più registrate ulteriori crisi.
L'epilessia
benigna occipitale a insorgenza precoce (Early-onset benign
occipital seizure syndrome) va distinta dalla più rara e
tardiva epilessia idiopatica occipitale con sintomi visivi diurni (3)
e rappresenta verosimilmente la più frequente causa di crisi
epilettiche focali nell'infanzia, dopo l'epilessia rolandica (4).
I bambini
colpiti anche in questo caso sono per definizione normali dal punto
di vista neurologico. L'età di incidenza varia fra 1 e 10
anni, ma è quasi sempre sotto i 5 anni (5).
Caratteristica
risulta la durata, spesso prolungata, della crisi d'esordio che può
talora mimare uno stato epilettico evocante una eziologia grave (6).
Ciononostante, la crisi ha scarsa tendenza a ripetersi e spesso
rimane unica (30%). I sintomi compaiono per lo più nel sonno o
al risveglio e spesso presentano un tipico esordio graduale e
fluttuante caratterizzato da: perdita del contatto e sguardo fisso,
deviazione tonica dello sguardo e/o del capo, ipotonia;
frequentemente si associano sintomi vegetativi tra cui nausea,
vomito, enuresi, tosse, cefalea. Solo raramente, la crisi sfocia in
fenomeni clonici unilaterali o anche generalizzati, che sono invece
più tipici dell'epilessia rolandica (v. tabella).
Epilessia
a punte rolandiche | Epilessia
benigna occipitale precoce | |
Età
d'esordio | 3-13
aa. (media 9) | 1-10
aa (media 4) |
Sesso
M | 60 % | 40 % |
Crisi | clonie
oro-facio-brachiali unilaterali disartria, scialorrea talora
sec.generalizzazione | deviazione
tonica capo/sguardo fluttuazione del contatto fenomeni vegetativi |
Frequenza
crisi | infrequenti,
talora a grappolo | rare,
spesso singola |
Risoluzione
clinica | <
13 anni | <
12 anni |
EEG
intercritico | EEG
intercritico punte lente centro-temporali talora controlaterali
indipendenti parossismi occipitali uni/bilaterali talora
coesistenti centro-temporali | parossismi
occipitali uni/bilaterali talora coesistenti centro-temporali |
Dal punto
di vista EEG i tracciati intercritici possono evidenziare parossismi
tipo punta, punta-onda o anche onde lente occipitali uni- o
bilaterali che si attenuano con l'apertura degli occhi. L'andamento
nel tempo di tali anomalie parossistiche è variabile, come
tipico in generale delle epilessie focali idiopatiche, potendo in
momenti diversi essere rappresentate in modo più o meno ricco
e/o alterno sui due emisferi. In una discreta percentuale di casi
possono coesistere o comparire più tardivamente parossismi
centro-temporali tipici dell'epilessia rolandica, le cui
manifestazioni cliniche possono in qualche caso subentrare
successivamente (7).
L'evoluzione
è generalmente benigna, con rare recidive e risoluzione
spontanea senza reliquati entro l'età della scuola
elementare. Nonostante non ci siano segnalazioni di disturbi
cognitivi in questa specifica forma, è comunque bene seguire
questo aspetto come consigliato nelle altre forme di epilessia focale
idiopatica. (8,9).
La
comparsa di una prima crisi epilettica è un evento in genere
molto stressante per i genitori, non solo per la drammaticità
della presentazione, ma anche per la preoccupazione che evoca sulle
possibili cause. La possibilità che il pediatra, che sempre
più spesso oggi è il primo medico ad affrontare la
situazione, sappia comunicare in modo equilibrato qualche elemento di
orientamento diagnostico e prognostico fin dall'inizio è
sicuramente di grande sostegno alla famiglia.
Questo è
particolarmente importante in quanto in un bambino sano una prima
crisi spontanea, specie se occorsa nel sonno, ha buone probabilità
di essere causata da una epilessia focale benigna (2),
tanto che secondo la Società Americana di Neurologia tali
quadri, quando tipici, non richiedono necessariamente un esame
neuroradiologico all'esordio (1).
La conoscenza degli specifici aspetti di presentazione clinica da
parte del pediatra può risultare inoltre utile anche nella
discussione con il neurologo consulente (che spesso può non
essere un esperto della patologia epilettica infantile) per orientare
l'approccio diagnostico-terapeutico.
L'epilessia
rolandica, più frequente e meglio conosciuta, nella maggior
parte dei casi è caratterizzata da crisi cloniche focali per
lo più fugaci, anche se talvolta possono secondariamente
generalizzare, con comparsa nel sonno, frequenza sporadica ed
evoluzione verso la spontanea risoluzione, tanto che nella maggior
parte dei casi non necessita di un trattamento farmacologico.
La
conoscenza dell'epilessia occipitale benigna precoce, le cui
manifestazioni tendono ad essere piuttosto stereotipate, consente
l'orientamento verso questa sindrome benigna già
dall'esordio, nonostante questo possa essere piuttosto spettacolare
e preoccupante, come abbiamo visto in particolare nel caso 3.
Naturalmente,
sebbene il quadro elettro-clinico sia normalmente molto
caratteristico, è comunque opportuno escludere altre
situazioni favorenti le crisi occipitali come la celiachia (con o
senza calcificazioni cerebrali) (10)
o lesioni epilettogene organiche.
1)
Freeman JM. Less testing is needed in the emergency room after a
first afebrile seizure. Pediatrics 2003;111:194-196
2) Hirtz
D, Ashwal S et al. Practice parameter: Evaluating a first nonfebrile
seizure in children. Neurology 2000;55:616-623
3)
Gastaut H. A new type of epilepsy: benign partial epilepsy of
childhood with occopital spike-waves. Clinical Electroencephalography
1982;13:13-22
4)
Panayiotopoulos CP. Early-onset benign childhood occipital seizure
susceptibility syndrome: a syndrome to recognize. Epilepsia
1999;40:621-630
5) Ferrie
CD et al. Early-onset benign occipital seizure susceptibility
syndrome. Epilepsia 1997;38:285-293
6) Nahum
E, Kivity S et al. Benign occipital epilepsy mimicking a catastrophic
intracranial event. Pediatric Emergency Care 2001;17:196-198
7)
Caraballo R, Cersosimo R, Fejerman N. Idiopathic partial epilepsies
with rolandic and occipital spikes appearing in the same children.
Journal of Epilepsy 1998;11:261-264
8)
Tenenbaum SN et al. Continuous spike-waves and dementia in childhood
epilepsy with occipital paroxysms. J Epilepsy 1997;10:139-145
9) Yung
AW, et al. Cognitive and behavioural problems in children with
centrotemporal spikes. Pediatr Neurol 2000;23:391-395
10) Gobbi
G, Bouquet F et al. Coeliac disease, epilepsy, and cerebral
calcifications. Lancet 1992;340:439-43
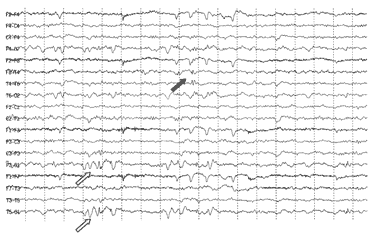
Fig.1:
anomalie lente aguzze occipitali sinistre (freccia vuota) coesistenti
con sporadiche punte lente trifasiche
centrotemporali
controlaterali (freccia piena)
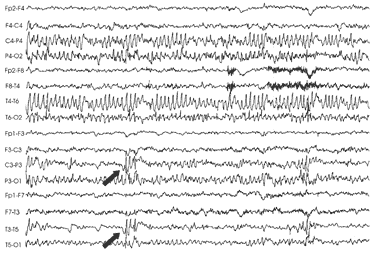
Fig
2: onde lente aguzze subcontinue parietooccipitali a destra e
triplette di punteonda
parietooccipitali
a sinistra (freccia piena)
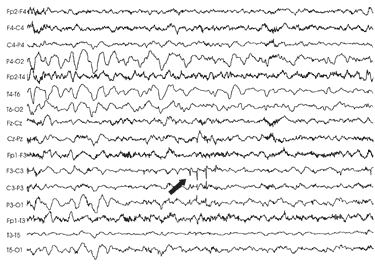
Fig
3: attività lenta delta occipitale prevalente a destra, punte
rolandiche a sinistra (freccia piena)
Vuoi citare questo contributo?