Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Febbraio 2025 - Volume XXVIII - numero 2
M&B Pagine Elettroniche
I Poster degli specializzandi
Leishmania e linfoistiocitosi emofagocitica (HLH)
1Dipartimento di Neuroscienze, Riabilitazione, Oftalmologia, Genetica e Scienze Materno-Infantili (DINOGMI),
Università di Genova
2UOC Ematologia Pediatrica, IRCCS Istituto Giannina Gaslini, Genova
Indirizzo per corrispondenza: annabratta6@gmail.com
Introduzione
La linfoistiocitosi emofagocitica (HLH) è una condizione rara, ma potenzialmente letale, che deve essere precocemente riconosciuta per evitare complicanze a lungo termine. Deriva da una persistente attivazione e da una intensa proliferazione dei linfociti T citotossici e delle cellule natural killer (NK), con produzione di grandi quantità di citochine che portano allattivazione degli istiociti (macrofagi e cellule dendritiche), che a loro volta causano la formazione di infiltrati tissutali e iperproduzione di citochine pro-infiammatorie.
Le HLH possono essere distinte in primitive (su base genetica) o secondarie a infezioni (tra le più comuni ricordiamo EBV, varicella, Leishmania spp.) o a farmaci. Nel caso in cui il quadro sia secondario a patologie reumatologiche, si parla di sindrome da attivazione macrofagica (MAS). La diagnosi si basa sui criteri dellHistiocyte Society del 2004: si può parlare di HLH in presenza di genetica patologica o nel caso in cui siano presenti almeno 5 delle seguenti condizioni:
- febbre
- splenomegalia
- citopenia di almeno due linee cellulari (emoglobina < 9 g/dl (nei neonati: < 10 g/dl); piastrine < 100.000/ml; neutrofili < 1000/ml)
- ipertrigliceridemia e/o ipofibrinogenemia (trigliceridi a digiuno ≥ 265 mg/dl; fibrinogeno ≤ 1,5 g/dl)
- emofagocitosi nel midollo osseo o nella milza o nei linfonodi
- bassa o assente attività delle cellule NK
- ferritina ≥ 500 ug/l
- CD25 solubile (sIL2Rx) ≥ 2400 U/I.
Caso clinico
Il nostro paziente, un lattante di 5 mesi, veniva ricoverato presso altro presidio ospedaliero, per sindrome iperinfiammatoria in corso di definizione diagnostica, con febbre persistente da circa 10 giorni non responsiva a terapia antibiotica, innalzamento degli indici di flogosi (PCR 22,8 mg/dl, ferritina 1.009 ng/ml) e splenomegalia. Dopo aver escluso possibili cause di HLH secondaria a processi infettivi (sierologie virali negative, ricerca di Leishmania spp. su sangue midollare negativa) e aver escluso presenza di cloni neoplastici alla valutazione midollare, veniva avviata terapia steroidea per os.
Per scadimento delle condizioni cliniche generali e persistenza di esami ematochimici compatibili con il perdurare di processi autoinfiammatori, il piccolo veniva trasferito presso il nostro ospedale per sospetta HLH (verosimilmente su base genetica), in considerazione delletà, della gravità e della resistenza alla terapia steroidea di prima linea.
Allingresso in reparto, previa ripetizione di agoaspirato midollare, si procedeva a potenziamento della terapia immunosoppressiva e antinfiammatoria; inoltre, per la gravità del quadro, si procedeva allesecuzione di indagini genetiche urgenti, che consentivano di escludere mutazioni di geni associati alla HLH familiare o a disimmunità. Si escludevano nuovamente tutte le cause secondarie di HLH. Dal punto di vista terapeutico, si associava alla terapia con desametasone inizialmente anakinra e ciclosporina (sospesa per tossicità neurologica), e successivamente etoposide.
Per la persistenza e per lingravescenza dellinfiammazione, si procedeva alla somministrazione di emapalumab, anticorpo monoclonale anti-interferon-gamma indicato in pazienti con HLH ricorrente progressiva o refrattaria ad altri trattamenti. Nonostante tale terapia, si assisteva a un progressivo peggioramento del quadro clinico, suggestivo di immunodeficit primitivo anche in assenza di una documentata mutazione genetica specifica. Si riteneva quindi il paziente potenzialmente candidabile a trapianto allogenico urgente di cellule staminali ematopoietiche.
Cautelativamente, si procedeva allattuazione di una nuova valutazione midollare (terza in totale), su cui si metteva in evidenza in morfologia la presenza inequivocabile di numerosi elementi parassitari (Figura) compatibili con la diagnosi di leishmaniosi, diagnosi che veniva confermata anche in sierologica e in biologia molecolare.
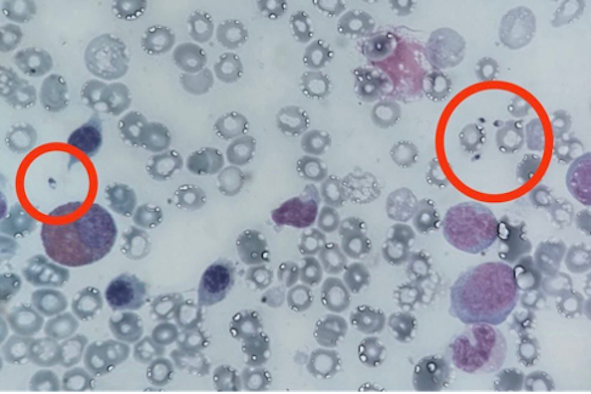
ll trattamento specifico delle leishmaniosi viscerali con amfotericina B liposomiale e il contestuale graduale scalo della terapia immunosoppressiva consentivano il miglioramento graduale delle condizioni cliniche generali e degli indici infiammatori; al fine di una completa normalizzazione della citopenia si rendeva necessaria lassociazione con fattori di crescita (GCSF/EPO).
Dopo un anno dallevento, il bambino è in ottime condizioni cliniche generali, ha 16 mesi e, finalmente, presenta emocromo nella norma, ma persiste una modesta splenomegalia e un lieve incremento delle transaminasi.
Linterpretazione di questo caso ha certamente rappresentato una sfida. Lesperienza nellambito della HLH ha consentito di rivalutare in modo critico il percorso diagnostico, alla luce di una clinica poco compatibile con una forma primitiva vista la risposta transitoria e insufficiente al trattamento immunosoppressivo, e si è quindi potuto giungere alla diagnosi definitiva di parassitosi. Lerrore diagnostico e lattuazione del trapianto di cellule staminali emopoietiche avrebbe posto a rischio di vita il piccolo paziente.
Bibliografia di riferimento
- Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124-31. DOI: 10.1002/pbc.21039.
- Jenkins MR, Griffiths GM. The synapse and cytolytic machinery of cytotoxic T cells. Curr Opin Immunol 2010;22(3):308-13. DOI: 10.1016/j.coi.2010.02.008.
- Lepore L (a cura di). Reumatologia per il Pediatra - Seconda Edizione. Medico e Bambino sas, Trieste. 2019.
- Perez N, Virelizier JL, Arenzana-Seisdedos F, Fischer A, Griscelli C. Impaired natural killer activity in lymphohistiocytosis syndrome. J Pediatr 1984;104(4):569-73. DOI: 10.1016/s0022-3476(84)80549-1.
Vuoi citare questo contributo?