Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Novembre 2004 - Volume VII - numero 10
M&B Pagine Elettroniche
Seminari degli specializzandi
Maturazione
del sistema immunitario e difetti congeniti
Tutor:
Giacomet Vania
Clinica
Pediatrica, Ospedale L. Sacco, Milano
Clinica
diretta da Prof. di Natale Berardo
Indirizzo
per la corrispondenza: alessandradepalma@libero.it
GENERALITA'
SUL SISTEMA IMMUNITARIO
Cosa si
intende per sistema immunitario ed immunità ?
Definiamo
Immunità la risposta del nostro organismo verso sostanze
estranee, indipendentemente dalle conseguenze fisiologiche e
patologiche che possono derivare da tale processo. L'insieme delle
cellule e delle molecole coinvolte in questa risposta prende il nome
di Sistema immunitario, complesso deputato al riconoscimento del self
dal non-self, e la distruzione di quest'ultimo al fine di mantenere
integra l'identità (= omeostasi= condizione di salute).
L'immunità
è divisibile in due branche principali: Naturale eAcquisita.
La prima
è definita naturale in quanto composta da meccanismi di
difesa preesistenti all'esposizione agli agenti microbici o alle
macromolecole estranee, la cui attività peraltro non viene
potenziata da tale esposizione. Altra caratteristica è che non
è in grado di discriminare, se non in modo grossolano, tra le
diverse sostanze estranee. Le strutture che costituiscono
l'immunità naturale sono: le barriere fisiche, i
fagociti, gli eosinofili, le cellule Natural Killer ed infine
molecole sia circolanti (il complemento) che solubili (le citochine).
Al
contrario, l'immunità acquisita è
caratterizzata da: specificità, diversità, memoria e
autoregolazione; il che permette alle strutture che la compongono di
potenziarsi col susseguirsi delle esposizioni al non-self e quindi di
“acquisire” una maggiore capacità difensiva. L'immunità
acquisita si divide a sua volta in Umorale e Cellulare.
L'umorale
è messa in opera da Linfociti B i quali sono deputati alla
produzione di anticorpi in grado di difendere l'organismo da
tossine e microrganismi extracellulari. La parte cellulare invece è
costituita dai Linfociti T che si occupano della risposta contro i
virus e i microrganismi intracellulari, ma che sono anche in grado di
potenziare e maturare le risposte umorali e naturale.
Tabella
1. IMMUNITA' NATURALE E SPECIFICA
NATURALE | SPECIFICA | |
BARRIERE
FISICHE | Cute,
mucose | Sistema
immune cutaneo e associato alle mucose; Anticorpi nelle
secrezioni mucose |
MOLECOLE
CIRCOLANTI | Complemento | Anticorpi |
CELLULE | Fagociti
(monociti, macrofagi, neutrofili)
NK | Linfociti |
MEDIATORI
SOLUBILI | Citochine
di derivazione macrofagica | Citochine
di derivazione linfocitaria |
Il
bersaglio di questo imponente “scudo difensivo” dell'organismo
è l'Antigene che viene definito come una qualsiasi
molecola in grado di indurre, una volta riconosciuta dal S.I., la
formazione di anticorpi (che riconoscono gli antigeni solubili)
oppure di linfociti citotossici (che riconoscono gli antigeni sulla
superficie delle cellule accessorie APC) atti ad eliminarlo.
Ilinfociti sono morfologicamente uguali; si distinguono sia dal
punto di vista della funzionalità, sia dalla presenza di
marcatori di superficie (proteine). Nascono nel midollo osseo delle
ossa piatte per poi seguire due destini differenti: i linfociti T
migrano per via ematica nel timo dove poi giungono a maturazione,
mentre i linfociti B maturano in fase precoce nel midollo osseo.
Entrambe le popolazioni di linfociti migrano negli organi linfatici
periferici (linfonodi, placche di Peyer, tessuto linfatico cute e
mucose).
I
Linfociti di classe B, a contatto con l'Ag diventano Plasmacellule
in grado di secernere gli anticorpi circolanti nel siero.
I
Linfociti T sono invece suddivisi in Citotossici, deputati alla lisi
dell'Agente estraneo, o in Helper deputati alla produzione di
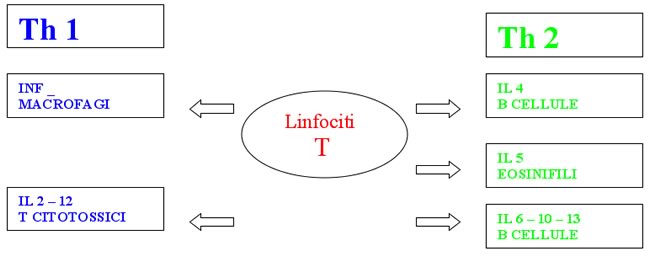
citochine. Questi ultimi a loro volta si differenziano in Th1,
attivanti i linfociti T e in Th2 attivanti i linfociti B. Esiste
inoltre una classe di linfociti T regolatori, costituita dai Th3, in
grado di favorire la produzione di IgA, con proprietà
soppressive sui Th1 e Th2 e rilascianti il TGF-_ e dai Tr1, in grado
di rilasciare oltre al TGF-_ (trasformino growth factor) anche IL-10.
Data l'importanza del TGF-_ nell'immunoregolazione e nella
tolleranza pare anche evidente la responsabilità di tale
classe di linfociti in alcune immunodisregolazioni su base genetica.
I
linfociti T sono il centro direzionale della risposta immunitaria. La
ricircolazione linfatica facilita il contatto tra antigene e
linfocita, dal momento che i linfociti continuamente passano dal
sangue ai tessuti, e da questi grazie alla linfa giungono ai
linfonodi, per poi ritornare nel circolo sanguigno grazie al dotto
toracico
Figura
1. CLASSI DI LINFOCITI T.
Glianticorpi sono glicoproteine presenti in tre diverse forme:
- legati alla membrana dei linfociti B;
- solubili: nel siero ed in fluidi secretori;
- legati a NK e fagociti.
Gli
anticorpi svolgono funzioni di strutture recettoriali, sono in grado
di legare e neutralizzare gli antigeni, attivano il complemento,
favoriscono il meccanismo di fagocitosi grazie al fenomeno di
opsonizzazione dell'antigene ed infine sono in grado di attivare
l'ADCC, cioè la lisi di cellule tumorali o infette mediata
dalle cellule NK.
Tabella
2. CLASSIFICAZIONE Ig
CLASSE
di Ig | FUNZIONE |
Ig
G_M | Attivazione
del complemento |
Ig G | Opsonizzazione |
Ig G
(per NK)
Ig
E_A (per eosinofili) | ADCC
(citotossicità cellulare Ab dipendente) |
Ig E | Ipersensibilità
immediata |
Ig A | Immunità
a livello delle mucose |
Ig G
materne
Ig A
nel latte umano | Immunità
neonatale |
Ig G | Circuiti
di inibizione delle risposte immuni |
Ilcomplemento è un sistema di proteine funzionalmente
collegate, che interagiscono tra loro a cascata, le cui funzioni
principali sono rappresentate da:
- Opsonizzazione
- Attivazione del processo infiammatorio
- Neutralizzazione degli ImmunoComplessi
- Mediatori della citolisi
Le
citochine sono un gruppo di proteine ormonali che regolano la
proliferazione, differenziazione, attivazione delle cellule nella
fase effettrice della risposta immune. Esse sono divise in 4 gruppi
in base alla loro funzione (anche se molte citochine possono svolgere
più di una funzione):
- Mediatori dell'immunità naturale (prodotte dai fagociti mononucleati)
- Regolatori di: attivazione, crescita e differenziazione linfocitaria
- Regolatori del processo infiammatorio
- Stimolanti la crescita e la differenziazione dei leucociti immaturi
SISTEMA
IMMUNITARIO: dal neonato alla completa maturazione
Le
diverse linee cellulari del sistema immunitario si sviluppano da
“cellule staminali totipotenti” a partire dalla 5° settimana
di gestazione. Esse sono distribuite attraverso i vari organi nel
sistema linfatico. I linfociti B e T, come già ricordato, sono
gli unici componenti del sistema immunitario che hanno capacità
di riconoscimento antigene-specifica; essi, infatti, sono
responsabili dell'immunità adattativa. I linfociti T si
sviluppano in differenti organi alle diverse epoche gestazionali:
- midollo osseo (8-10 settimane);
- timo (8 settimane);
- linfonodi (11 settimane);
- appendice (11 settimane);
- tonsille (14 settimane).
Figura
2. GENESI DEL SISTEMA IMMUNITARIO
Tratto
da: “Nelson” XVI edizione 2000

Attraverso
tali organi, il sistema linfatico è responsabile della
diffusione delle componenti del sistema immunitario nelle varie parti
del corpo.
Alla
nascita il neonato può usufruire di due tipi d'immunità:
“Attiva”, debole e non ancora in grado di proteggere in modo
adeguato il bambino dall'aggressione dei patogeni e “Passiva”,
composta da immunoglobuline acquisite dalla madre attraverso il
passaggio placentare. Tuttavia gli anticorpi materni non hanno una
trasmissione verticale equa, infatti alla nascita i neonati sono in
grado di rispondere alle aggressioni da parte dei virus e degli
organismi Gram positivi, ma non contro i Gram negativi. Questi ultimi
sono però i responsabili di numerose patologie che si possono
manifestare proprio nel periodo neonatale, ad esempio: la Gonorrea,
la Pertosse, la Salmonella, la Shigella, il Colera o l'infezione da
E. Coli.
L'immunitàpassiva comunque è un valido supporto alla scarsità
delle difese endogene, sotto forma di:
- alti livelli di IgG passate attraverso la placenta a partire dalla XII settimana di gestazione che diminuiscono progressivamente nei primi mesi di vita;
- IgA materne secrete nel colostro e nel latte materno.
L'immunitàattiva invece è caratterizzata, rispetto all'età
adulta, da: ridotta produzione di citochine, minore attività
del complemento e significativa riduzione dei suoi fattori, pool
midollare di neutrofili ridotto, risposta dei T-linfociti agli
antigeni più lenta e inoltre una produzione anticorpale
rallentata; tutti questi fattori contribuiscono a rendere il periodo
neonatale un lasso di tempo ad alto rischio infettivo durante il
quale i processi infetti sia virali che batterici decorrono con
maggior gravità.
I neonati
iniziano a sintetizzare anticorpi di classe IgM immediatamente dopo
la nascita, in quota sempre crescente, a seguito della notevole
stimolazione antigenica che deriva dall'ambiente esterno. I
prematuri sembrano essere capaci di tale risposta in maniera del
tutto simile ai nati a termine. Dopo circa 6 giorni dalla nascita i
livelli di IgM subiscono un rapido incremento che continua fino al
raggiungimento dei livelli simile all'adulto approssimativamente al
compimento del primo anno di vita.
Per ciò
che concerne la quota di IgG, mentre quelle materne gradualmente
diminuiscono nell'arco dei primi 6-8 mesi, quelle prodotte dal
bambino tendono progressivamente ad aumentare fino a stabilizzarsi
sulla concentrazione dell'adulto ai 7-8 anni d'età. Le
IgG1 e le IgG4 sono le prime a raggiungere i livelli adulti seguite
dalle IgG3 (verso i 10 anni) e le IgG2 (verso i 12 anni). In ogni
modo i bambini sani non possono produrre anticorpi contro antigeni
polisaccaridici solitamente fino ai 2 anni d'età, a meno che
il polisaccaride non sia coniugato con una proteina carrier, come nel
caso dei vaccini coniugati Haemophilus influenzae di tipo b (Hib), e
PVC; inoltre, sotto i due anni d'età gli Ag T-indipendenti
sono poco immunogeni. Tutto ciò comporta una maggiore
suscettibilità alle infezioni provocate da batteri la cui
capsula sia costituita da polisaccaridi. Lo sviluppo del sistema
immunitario si completa nei primi anni di vita in seguito
all'interazione del bambino con l'ambiente e alle stimolazioni
antigeniche cui è sottoposto. Tuttavia la varietà della
stimolazione antigenica e la cooperazione tra linfociti B e T
contribuiscono ad una completa maturazione del sistema immunitario
sin dal primo anno di vita.
Tabella
3. LINFOCITI: VALORI NORMALI PER L'ETÀ
Tratto da: “Interpretation of the complete blood count.”
Pediatrics clinics of North America 1996
Età | Linfociti
x1000/uL | %
su GB |
Nascita | 2.0-11.0 | 31% |
1
settimana | 2.0-17.0 | 41% |
1
mese | 2.5-16.5 | 56% |
6
mesi | 4.0-13.5 | 61% |
12
mesi | 4.0-10.5 | 61% |
4
anni | 2.0-8.0 | 50% |
8
anni | 1.5-6.8 | 39% |
21
anni | 1.0-4.8 | 34% |
Tabella
4. LIVELLI SIERICI NORMALI DI IMMUNOGLOBULINE (+/- 2DS)
Età | IgG | IgA | IgM
(mg/dl) |
Cordone
Ombelicale | 1112
(862
- 1434) | Non
dosabili | 9
(5-14) |
1-3
mesi | 468
(231
- 497) | 24
(8 -
74) | 74
(26 -
210) |
4-6
mesi | 434
(222-846) | 20
(6-60) | 62
(28-39) |
7-12
mesi | 569
(351-919) | 29
(10-85) | 89
(38-204) |
13-24
mesi | 801
(264-1509) | 54
(17-178) | 128
(48-337) |
2-3
anni | 889
(462-1710) | 68
(27-173) | 126
(62-257) |
4-5
anni | 1117
(528-1959) | 98
(37-257) | 119
(49-292) |
6-8
anni | 1164
(633-1016) | 113
(41-315) | 121
(56-261) |
9-11
anni | 1164
(707-1919) | 127
(60-270) | 129
(61-276) |
12-16
anni | 1105
(640-1909) | 136
(61-301) | 132
(59-297) |
QUANDO
SOSPETTARE UN'IMMUNODEFICIENZA?
Anamnesi
Un'
accurata indagine anamnestica è fondamentale nel discriminare
quali soggetti possano far sospettare un'immunodeficienza,
necessitando, quindi, di indagini più approfondite.
È
importante indagare già dall'anamnesi familiare la presenza
d'infezioni severe, sepsi, immunodeficienze o morti precoci
(costruire albero genealogico).
Per ciò
che concerne la storia clinica del paziente, bisognerà
raccogliere informazioni dettagliate riguardo all'età
gestazionale, il peso alla nascita, il periodo neonatale e, la
risposta alle vaccinazioni.
Infine
molta attenzione va posta all'anamnesi patologica prossima e remota
indagando sulla natura, severità, sede e ricorrenze delle
infezioni (aumentata frequenza, gravità, durata prolungata,
recidive senza intervallo libero, aumentata dipendenza dagli
antibiotici, complicanze infettive gravi o inaspettate, infezioni da
parte di microrganismi insoliti, solitamente opportunisti).
Caratteristiche
cliniche associate ad immunodeficienza
Il
pediatra si trova spesso ad osservare bambini con infezioni
ricorrenti, tuttavia la maggioranza di questi non ha una patologia da
immunodeficienza. Si parla d'infezioni respiratorie ricorrenti
(IRR) nel caso di un bambino che nei primi anni di vita (1-6 anni),
sviluppa più di 6 infezioni/anno o più di
un'infezione/mese nel periodo autunno-inverno e che non abbia
condizioni patologiche sottostanti (anomalie congenite delle vie
aeree, fibrosi cistica, IDP, infezione da HIV, sindrome delle ciglia
immobili).
L'incidenza
di tale condizione è stimata intorno al 6% dei bambini
italiani. Essa è provocata da una fisiologica immaturità
del sistema immunitario su cui si inseriscono sia l'effetto
sfavorevole dell'impatto ambientale (socializzazione precoce,
esposizione al fumo passivo, inquinamento atmosferico), sia l'azione
immunodepressiva transitoria indotta dagli stessi agenti patogeni. E'
una condizione benigna ed autorisolventesi nel corso di 1-2 anni.
Dal punto
di vista clinico il quadro che si presenta è eterogeneo,
caratterizzato da infezioni aspecifiche delle alte vie respiratorie e
meno frequentemente delle basse vie respiratorie che non differiscono
per durata e gravità da quelle dei bambini con normale
incidenza di malattie delle vie respiratorie. Sono determinate per lo
più da virus (Rhinovirus, Virus parainfluenzali tipo 1,2,3,4 e
virus influenzali tipo A e B). Tra i batteri, anche se raramente
causa di IRR, nel 20% dei casi viene isolato lo Streptococco
_-emolitico gruppo A, (è possibile che un'elevata
percentuale sia tuttavia costituita da portatori sani) seguito a
distanza da Haemophilus Influenzae, Streptococcus Pneumoniae,
Moraxella Catarrhalis. La valutazione del sistema immunologico
dovrebbe essere intrapresa quindi solo in quei bambini con
manifestazioni cliniche specifiche di patologie del sistema
immunitario o con infezioni inusuali, croniche o ricorrenti.
Riportiamo
di seguito le condizioni cliniche che dovrebbero far sospettare la
presenza di un'immunodeficienza sottostante. Inoltre ricordiamo
come il tipo d'infezione fornisca dati importanti riguardo alla
qualità del difetto presente. (vedi tab. 4)
Dati
frequentemente presenti e altamente indicativi
- Infezioni ricorrenti, specie se politopiche
- Infezioni croniche
- Agenti microbici inusuali
- Incompleta risoluzione tra gli episodi infettivi o scarsa risposta alla terapia
Tabella
4. CORRELAZIONE TRA INFEZIONI E TIPO DI DEFICIT
PRINCIPALI
INFEZIONI | DEFICIT |
Infezioni
ricorrenti e/o gravi da batteri invasivi extracellulari
(Haemofilus, Stafilococco, Pneumococco, Streptococco,
Pseudomonas); infezioni virali e protozoarie persistenti
(Giardia).
In
particolare: otiti purulente, polmoniti,meningiti, sepsi. | DEFICIT
ANTICORPALI |
Infezioni
disseminate da agenti microbici intracellulari. | DEFICIT
DELL'IMMUNITÁ CELLULO-MEDIATA |
Infezioni
da agenti sia intra- che extracellulari | IMMUNODEFICIENZE
COMBINATE |
Infezioni
gravi politopiche (Candida, Streptococco, Stafilococco). | DEFICIT
DEI FAGOCITI |
Aumentata
incidenza di malattie autoimmuni;
Infezioni
da batteri piogeni. | DEFICIT
DEL COMPLEMENTO |
Dati
frequentemente presenti e moderatamente indicativi
- Lesioni cutanee (eczema, candidosi, rash, seborrea, alopecia, verruche)
- Diarrea cronica
- Ritardo di crescita
- Epatosplenomegalia
- Anomalie ematologiche
- Ascessi ricorrenti
- Osteomieliti ricorrenti
- Autoimmunità
- Difetti di sviluppo
Dati
associati con quadri specifici di immunodeficienza
- Atassia-teleangectasia (S. di Louis_Bar)
- Nanismo ad arti corti
- Endocrinopatie
- Albinismo parziale
- Tetania
- Trombocitopenia-eczemi (S. di Wiskott-Aldrich)
- Periodontite
SCREENING
IMMUNOLOGICO DEL BAMBINO CON IRR
Di fronte
ad un paziente con infezioni respiratorie ricorrenti è
opportuno indagare con degli esami comuni e poco costosi, ma che ci
possono fornire importanti informazioni.
- Emocromo completo con formula, VES.
- Test di screening per i difetti B-cellulari
- Dosaggio delle IgA, IgM e IgG;
- Isoemoagglutinine (anti-A, anti-B);
- Titolo anticorpale per tetano, difterite.
- Test di screening per i difetti T-cellulari
- Conta assoluta dei linfociti.
- Test di screening per i difetti di fagocitosi
- Conta assoluta dei neutrofili.
- Test di screening per il deficit del complemento
- Attività emolitica totale (CH 50);
- C3, C4.
TEST
DIAGNOSTICI AVANZATI
Nel caso
in cui dalla clinica e dagli esami di primo livello scaturisca il
sospetto diagnostico di immunodeficienza, ed a maggior ragione se vi
sono indicazioni riguardo la qualità del difetto presente, si
può procedere all'esecuzione di indagini più accurate
e specifiche.
Test
per lo studio del sistema anticorpale
- Valutazione della concentrazione ematica delle cellule B mediante citofluorimetria;
- Determinazione quantitativa delle immunoglobuline (IgD, IgE);
- Sottoclassi IgG;
- Risposte anticorpali specifiche (Valutazione del titolo anticorpale prima e dopo l'immunizzazione con tossoide tetanico, tossoide difterico, polisaccaride capsulato di H. influenzae.
- Dosaggio della concentrazione delle Ig nel siero ed, eventualmente, in altri liquidi e secreti corporei.
Test
di valutazione dell'immunità cellulo-mediata
- Test cutanei di ipersensibilità ritardata (Candida, Tossoide tetanico);
- Conta dei linfociti T totali con anticorpi monoclonali (CD3);
- Valutazione in citofluorimetria dei subset T cellulari (CD4 e CD8) (determinano la percentuale di helper e citotossici);
- Valutazione di antigeni di attivazione e di memoria dei linfociti T (HLA DR, CD45RA, CD45R0)
- Risposta proliferativa a mitogeni (PHA), antigeni e cellule allogeniche (reazione leucocitaria mista);
- Produzione di citochine.
Test
di valutazione della fagocitosi
- Valutazione morfologica delle cellule per escludere l'esistenza di anomalie nelle caratteristiche granulazioni;
- Test di adesione, chemiotassi, test per la valutazione della fagocitosi, riduzione del nitroblu di tetrazolio (NBT) sostituito da citometria a flusso della combustione respiratoria;
- Numero e funzione delle cellule natural killer (NK) (CD16 CD56).
Test
di valutazione del complemento
- Determinazioni di componenti specifici;
- Studio dell'espressione dei recettori cellulari per componenti del complemento (CR1 e CR3).
LE
IMMUNODEFICIENZE
Sono un
gruppo assai eterogeneo di affezioni caratterizzate da difetti di
sviluppo o funzionamento del Sistema Immunitario che si traducono in
un'insufficiente risposta.
Esse
possono causare non solo un'aumentata suscettibilità alle
infezioni, ma anche favorire l'insorgenza di patologie autoimmuni e
neoplasie. Da un punto di vista epidemiologico l'incidenza è
stata calcolata essere di 1/10.000 (escludendo il Deficit
asintomatico di IgA che ha un'incidenza di 20/10.000), con comparsa
preferenziale nel sesso maschile.
L'età
di comparsa può essere suddivisa come segue:
- 40% nel primo anno di vita;
- 40% nei primi 5 anni di vita;
- 15% nel sesto anno di vita;
- 5% nell'età adulta.
Le
immunodeficienze possono essere distinte in:
Immunodeficienze
primitive
(CONGENITE)
Espressione di un vero e proprio difetto genetico (soprattutto
X-linked), caratterizzate clinicamente da aumentata suscettibilità
alle infezioni che si manifesta precocemente nell'infanzia o che
può occasionalmente evidenziarsi in età più
avanzata.
Incidenza:
1/50.000 nati vivi.
Immunodeficienze
secondarie
(ACQUISITE)
Si manifestano a seguito di malnutrizione, neoplasie, situazioni di
stress, dismetabolismi, trattamento con farmaci ad azione
immunosoppressiva o infezioni delle cellule immunocompetenti (HIV).
Noi
abbiamo dedicato particolare attenzione alle immunodeficienze
primitive, cercando di riassumere per ciascuna patologia nozioni di
pronto utilizzo al fine di avere una visione completa e più
chiara di queste sindromi di difficile diagnosi e gestione.
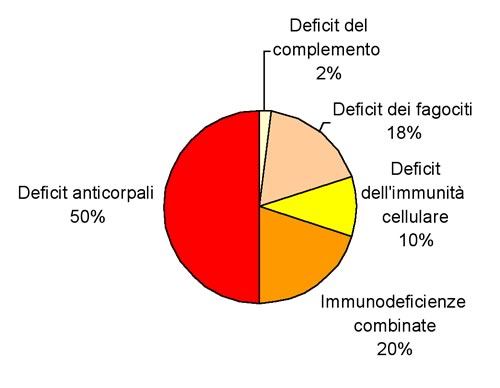
Iniziamo
dal proporre innanzitutto un prospetto che sia utile a dimostrare la
reale distribuzione di tali patologie sul territorio.
Figura
3. DISTRIBUZIONE DELLE IMMUNODEFICIENZE PRIMITIVE
Di
seguito, un cenno ai difetti immunologici più “frequenti”:
1)
DEFICIT ANTICORPALI
Deficit
di IgA
Condizione
trasmessa con carattere Autosomico Recessivo o sporadica, associata
al complesso maggiore di istocompatibilità di classe I o II.
Attualmente
in Italia la frequenza è stimata intorno a 1:600.
Gli esami
di laboratorio mostrano un'assenza totale o quasi (<5-10 mg/dL)
delle sole IgA sieriche e secretorie, con valori normali di IgG, IgM,
IgD e IgE e uno squilibrio tra IL5 ed IL6.
Sintomatologia:
Nel
50% dei casi i pazienti non presentano patologie rilevanti,
mentre
nel restante 50% si possono riscontrare:
- infezioni respiratorie ricorrenti gravità lieve-moderata, raramente polmoniti ricorrenti e bronchiectasie
- infezioni enteriche (diarrea da Giardia)
- allergie
- celiachia
- ritardo mentale
- patologie autoimmuni (LES, AR, anemia emolitica, dermatomiosite)
- neoplasie (linfomi intestinali)
Diagnosi
La
diagnosi si basa quindi sul dosaggio IgA e delle altre
Immunoglobuline.
La
prognosi dipende dalla patologia associata; per quel che riguarda la
terapia non esiste terapia specifica, ma solo sintomatica.
Agammaglobulinemia
X-linked (S. di Bruton)
I
pazienti con questo tipo di immunodeficienza primaria presentano un
grave difetto in numero e funzione dei linfociti B che esita in una
grave ipogammaglobulinemia.
A livello
genico l'anomalia risiede nella codificazione della tirosin-kinasi
di Bruton (espressa in dosi elevate in tutta la linea B-cellulare),
che esita nel blocco della maturazione dei linfociti B. Gli esami di
laboratorio evidenziano: IgG di solito < 200 mg/dL, assenza di IgA
e IgM , IgD, IgE, assenza quasi completa di linfociti B circolanti
(CD19 < 2%).
La
sintomatologia ha inizio dopo il 6° mese di vita in concomitanza
con la scomparsa degli Ab materni. La clinica può essere
caratterizzata da:
Sintomatologia:
- Infezioni respiratorie ricorrenti, gravi e recidivanti, resistenti alla terapia;
- Sinusiti, otiti, bronchiectasie causate da Pneumococco, Haemophilus, Stafilococchi, Pseudomonas;
- Diarrea da Giardia Lambia;
- Artriti da Mycoplasma;
- Fenomeni autoimmuni: anemia perniciosa, amiloidosi, AR, leucemie.
La
diagnosi si basa su: sesso maschile, anamnesi familiare, dosaggio
delle Immunoglobuline, carenza tessuto linfatico faringeo e assenza
di risposta anticorpale ai più comuni antigeni.
Prognosi:
La
prognosi dipende dalla terapia intrapresa, consistente in infusioni
di g-globuline (0.4-0.6g/Kg) ogni 3-4 sett. per mantenere
IgG>500mg/dL, in associazione ad antibioticoterapia per le
infezioni intercorrenti.
2)
DEFICIT DELL'IMMUNITA' CELLULO – MEDIATA
Sindrome
di Di George
Patologia
X-Linked o autosomica dominante (delezione 22q11), ma più
spesso sporadica.
E'
caratterizzata da insufficiente sviluppo del III-IV arco branchiale
con ipoplasia del timo e delle paratiroidi, anomalie cardiache
(tronco comune) e facciali (micrognazia, ipertelorismo, inserzione
bassa delle orecchie e frenulo nasale corto).
Sintomatologia:
L'esordio
si verifica solitamente a pochi giorni dalla nascita con ipocalcemia,
cardiopatia e facies tipica. La gravità clinica varia da
parziale linfopenia con moderata immunodeficienza fino al decesso
entro 2aa di vita.
La
terapia si avvale di: Ca + vitD e ormoni timici: nei casi in cui
l'immunodeficienza sia completa si deve ricorrere al trapianto di
midollo osseo HLA identico, non frazionato.
3)
DEFICIT COMBINATI
Severe
combined immunodeficiency (SCID)
Condizione
a carattere autosomico recessivo con diminuizione di T e B cell o
X-Linked con sola diminuizione di T cell e B cell normali, ma non
funzionanti per mancata cooperazione con i T. L'incidenza è
stimata intorno a 1:100.000.
Ne
esistono numerose varianti, ad es: difetto dei geni RAG e ARTEMIS,
difetto del gene dell'enzima adenosin-deaminasi (ADA), difetto
della proteina Z-attivante (ZAP), in caso di SCID a trasmissione
X-linked (X-SCID) è presente un difetto del gene IL-2R-gamma.
Tra le
immunodeficienze combinate ne esiste una variante con ipereosinofilia
ed elevazione delle IgE sieriche, la sindrome di Omenn: è una
condizione fatale, ereditaria autosomica recessiva, con profonda
suscettibilità alle infezioni con infiltrati T-cellulari in
cute, intestino, fegato e milza.
Clinicamente
le SCID si possono presentare con:
- infezioni precoci nei primi mesi di vita
- polmonite interstiziale
- diarrea
- candidiasi orale intrattabile
- infezioni da germi intracellulari
- reazioni trasfusionali importanti
La
diagnosi viene posta clinicamente dall'assenza di linfonodi
palpabili e di tonsille visibili,
ed
avvalorata dall'assenza dell'ombra timica all'Rx torace,
linfopenia all'emocromo e dalla tipizzazione linfocitaria.
La
prognosi è infausta con morte entro 1-2 aa per infezioni da
opportunisti e virus.
l'approccio
terapeutico si avvale di trapianto di M.O. eterologo compatibile da
fratello
Importante
ricordare che questi pazienti possono essere soggetti GRAVI
CONSEGUENZE se sottoposti a somministrazione di VACCINI VIVI
4)
DEFICIT DEI FAGOCITI
Malattia
granulomatosa cronica
Malattia
ereditaria rara, X-Linked, dovuta ad un deficit di NADPHossidasi.
L'esordio è spesso nei primi mesi di vita, più
raramente nell'età adulta.
Sintomatologia:
Clinicamente
si possono avere:
- Infezioni ricorrenti e gravi causate da germi catalasi positivi (Stafilococcus, Serratia, Aspergillo), miceti e batteri saprofiti.
- Granulomatosi con necrosi cutanea
- Osteomieliti
- Epatiti con epatomegalia
- Sepsi generalizzata
Diagnosi:
La
diagnosi viene posta dal test NBT e dal test di ossidazione della
diidrorodamina(DHR).
Terapia:
La
terapia si avvale di antibiotici aggressivi e prolungati, antifungini
± INF_ e nei casi più gravi da trapianto di M.O.
Deficit
di adesione leucocitaria (LAD)
Si
tratta di rari disordini autosomici recessivi della funzione
leucocitaria; ne esistono 2 tipi LAD-1 e LAD-2 . La LAD-1 colpisce
circa 1:1.000.000 ed è caratterizzata da infezioni batteriche
e fungine recidivanti e da ridotta risposta infiammatoria, nonostante
la presenza di neutrofilia molto spiccata.
Clinicamente
possono manifestarsi con:
- Infezioni ricorrenti batteriche di cute, bocca, tratto respiratorio, mucosa intestinale.
- Ritardo della caduta ed infezione del moncone ombelicale
- Gravi gengiviti con perdita precoce della dentizione decidua e poi permanente
La
diagnosi viene fatta mediante la dimostrazione alla citometria di
flusso di un deficit molecolare (CD 11/CD 18) responsabile
dell'alterazioni della chemiotassi dei neutrofili.
La
terapia si avvale dell'utilizzo di immunosoppressori e nei casi
gravi (completa assenza di molecole di adesione) si ricorre al
trapianto di midollo osseo allogenico.
Sindrome
di Job (iper IgE)
Sindrome
a trasmissione autosomica dominante a penetranza incompleta,
caratterizzata da livelli marcatamente elevati di IgE nel siero
(>2000 UI/ml) con eosinofilia tissutale e periferica e difetti
chemiotattici intermittenti.
L'esordio
è solitamente precoce nell'infanzia.
Sintomatologia:
Clinicamente
può manifestarsi sotto forma di:
- Infezioni cutanee e respiratorie, prevalentemente stafilococciche (polmoniti con formazione di pneumatoceli, mastoiditi; infezioni osteo-articolari meno frequenti)
- Eczema cronico
- Alterazioni facciali e scheletriche
Dagnosi:
La
diagnosi è data da aumento delle IgE, IgD moderatamente
aumentate e chemiotassi alterata.
Terapia:
La
terapia si avvale di un uso cronico di antibiotici.
Riferimenti
bibliografici
- Walters MC, Abelson HT. Interptretation of the complete blood count. Pediatrics clinics of North America 1996;43:599-617.
- Sterkers G. Immune defenses of newborn infants. Rev Prat.1991;41:1341-1344.
- Durandy A. Development of specific immunity in prenatal life. Arch Pediatrics 2001;8:979-985.
- Hayward AR. The human fetus and newborn: development of the immune response. Birth Defects Orig Art Ser. 1983;19:289-94.
- Quie PG. Antimicrobical defenses in the neonate. Semi Perinatol 1990; 14(4 suppl 1):2-9.
- Goldblatt D. Immunisation and the maturation of infant immune response. Dev Biol Stand. 1998;95:125-132.
- Millet V, Lacroze V, Bodiou AC, Dubus JC, D'Ercole C, Unal D. Ontogeny of the immune system.Arch Pediatr 1999;6 suppl 1:14S-19S.
- Muzyka BC. Host factors affecting disease transmission. Dent Clin North America 1996;40(2):263-275.
- Babu KS, Arshad SH. The role of allergy in the development of airway inflammation in children. Paediatr Respiratory Review 2003;4(1):40-46.
- Nelson. Textbook of Pediatrics. 16th ed. Philadelphia. 2000.
- Smith K.J, Skelton H. Common variable immunodeficiency treated with a recombinant human IgG, tumour necrosis factor-alpha receptor fusion protein. British Journal of Dermatology 2001; 144: 597-600.
- http.//www lark farm.com/AP/immune.htm: Development of the immune system. 2004.
- Ferguson AC, Cheung SS. Modulation of immunoglobulin M and G synthesis by monocytes and T lymphocytes in the newborn infant. Journal of Pediatrics 1981;98(3):385-391.
- Wolach B, Carmi D, Gilboa S, Satar M, Segal S, Dolfin T, Schlesinge. Some aspect s of the humoral immunity and the phagogytical function in newborn infants. Isr J Med Sci. 1994;30(5-6):331-335.
- Burgo G.R, Ugazio A.G. Immunologia e allergologia pediatrica. UPSA MEDICA.
- Abbas A.K, Lichtman A.H, Pober J.S. Cellular and molecular immunology. Philadelphia, 4th ed. 2000.
- Weiner HL. Induction and mechanism of action of transforming growth factor- (beta)- secreting Th3 regulatory cells. Immunol Rev 2001;182:207-214.
Vuoi citare questo contributo?