Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Dicembre 2024 - Volume XXVII - numero 10
M&B Pagine Elettroniche
I Poster degli specializzandi
Una diagnosi precoce di coartazione aortica e sindrome di Williams
1Scuola di Specializzazione in Pediatria, Università di Cagliari
2UO Terapia Intensiva Neonatale, Policlinico Universitario Duilio Casula, Monserrato, Università di Cagliari
Indirizzo per corrispondenza: siella.mazza@gmail.com
Giunge alla nostra attenzione una bambina nata da poche ore in un ospedale periferico, da cui è stata trasferita per soffio sistolico, ipoperfusione cutanea, polsi femorali deboli. Posta la diagnosi clinica ed ecocardiografica (Figura) di coartazione aortica serrata, è stata iniziata terapia ev con prostaglandina E1 (PGE1) e la piccola è stata avviata a intervento cardio-chirurgico di de-coartazione, eseguito in quinta giornata di vita.
Al controllo post dimissione la bambina presenta un iniziale restringimento del calibro dellarco dellaorta, ingravescente nei giorni successivi; viene inoltre riscontrato un restringimento dei rami polmonari periferici.
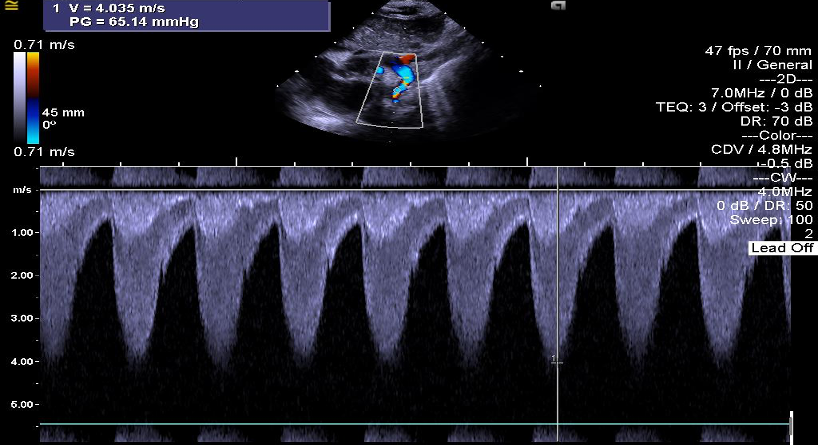
Le pressioni misurate allarto superiore destro e agli arti inferiori dimostrano uniniziale ri-coartazione aortica: pressione arteriosa (PA) AS 104/80; PA AAII 95/61 mmHg. Inoltre, si notano un edema palpebrale persistente, che inizialmente era stato associato al quadro di scompenso cardiaco neonatale, e un mento piccolo. Si pone pertanto il sospetto clinico di Sindrome di Williams e si avviano i test genetici.
Discussione
La Sindrome di Williams è caratterizzata da una microdelezione del braccio lungo del cromosoma 7, contenente anche il gene ELN che codifica per lelastina: questo comporta - con fenotipo variabile - una stenosi delle arterie di medio e grande calibro, associata a pareti vascolari inspessite e scarsamente elastiche. Oltre alle anomalie cardiovascolari e ai dismorfismi facciali, si riscontrano un profilo neuropsicologico, cognitivo e linguistico peculiare e anomalie del tessuto connettivo.
I pazienti con Sindrome di Williams e coartazione aortica devono essere sottoposti a monitoraggio clinico serrato, perché in letteratura sono riportati diversi seppur rari casi con aortopatie rapidamente progressive, che richiedono tempestivo riconoscimento e trattamento. In questi bambini la ri-coartazione sembra essere legata a unattiva ipertrofia delle cellule muscolari correlata allassenza di elastina a livello della parete vasale, più che da un fallimento della chirurgia nel mantenere un adeguato calibro del lume vasale.
È importante considerare, oltre allaumentato rischio di ri-coartazione, il rischio anestesiologico e il coinvolgimento multivasale (che può riguardare, oltre ad aorta e arterie polmonari, anche arterie renali, arterie mesenteriche e coronarie), che di per sé contribuiscono ad aumentare la mortalità.
Messaggi chiave
In caso di coartazione aortica, pensa alle sindromi genetiche potenzialmente associate, perché impattano sulla prognosi e sullatteggiamento terapeutico.
Bibliografia di riferimento
- Fornari, P. Cianci, A. Selicorni. La sindrome di Williams. Medico e Bambino 2018;37(2):111-113.
Vuoi citare questo contributo?