Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Maggio 2022 - Volume XXV - numero 5
M&B Pagine Elettroniche
I Poster degli specializzandi
Non ti scordar del tubulo: la malattia di Dent
Scuola di Specializzazione in Pediatria, IRCCS Materno-Infantile Burlo-Garofolo, Università di Trieste
Indirizzo per corrispondenza: simone.benvenuto2@icloud.com
Un ragazzo di 12 anni giunge allattenzione del nostro ambulatorio nefrologico per il riscontro accidentale di proteinuria in occasione di una visita medico-sportiva.
Il ragazzo non presenta familiarità per insufficienza renale né ematuria, e ad una prima valutazione lesame obiettivo è nella norma e la proteinuria risulta confermata, accompagnata da microematuria (stick urine: pH 6,5, PS 1030, prot 1+, GR 1+; al MO 30 GR/mm3 di morfologia mista).
Gli esami di laboratorio portati in visione mostrano una funzione renale normale (creatinina 0,65 mg/dl, eGFR sec. Schwartz 148 ml/min/1,72 m2), con rapporto proteinuria/creatininuria pari a 600 mg/g (vn < 200 mg/g). Nella norma anche lecografia addominale. Programmato un successivo controllo per approfondire il quadro, il ragazzo viene intanto valutato presso unaltra sede, dove lo studio delle urine delle 24 ore mostra una proteinuria in range non nefrosico (410 mg/24h) con calcio, fosforo, ossalati, acido urico e citrato urinari tutti nella norma; allecografia addominale riscontro di nefro-calcinosi a carico del rene sinistro.
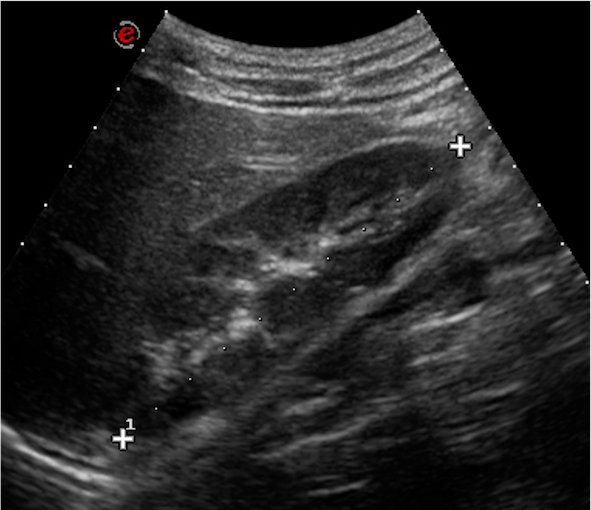
Dopo tre mesi il ragazzo ritorna presso il nostro ambulatorio: la proteinuria appare in netto peggioramento (1344 mg/24h), sempre accompagnata da microematuria, mentre si esclude una proteinuria ortostatica. Gli esami ematochimici eseguiti, comprensivi di emocromo, funzionalità renale ed epatica, assetto lipidico e proteico, complemento, indici di flogosi e autoanticorpi (ANA, anti-dsDNA, ANCA), risultano tutti nella norma. Allecografia delladdome confermati spot iperecogeni bilaterali come da nefrocalcinosi (Figura 1).
A indirizzare il sospetto diagnostico è però il rilievo di un valore marcatamente elevato di beta-2-microglobulina urinaria (> 50.000 ng/ml). Esclusa con una visita oculistica (risultata normale) una sindrome TINU (nefrite tubulo-interstiziale e uveite), consultiamo i genetisti per lavvio di indagini genetiche volte a confermare la diagnosi di una tubulopatia primitiva, e in particolare di una malattia di Dent: lanalisi dellesoma riscontrerà una mutazione nel gene CLCN5, patogenetica per malattia di Dent di tipo 1.
La malattia di Dent è una tubulopatia renale prossimale a trasmissione X-linked, causata da mutazioni nel gene CLCN5 (tipo 1) o OCRL1 (tipo 2). Clinicamente si caratterizza per proteinuria di basso peso molecolare, ipercalciuria (presente nel 75-90% dei casi) e nefrocalcinosi e/o nefrolitiasi; tipico anche il riscontro di micro- o macroematuria. Nei maschi affetti linsufficienza renale terminale arriva nel 30-80% dei casi in età adulta (tra i 30 e i 50 anni). Qualsiasi proteinuria, anche in range nefrosico (il 50% circa dei pazienti la svilupperà), dovrebbe indurre a escludere una malattia di Dent, specie se non accompagnata da edemi o ipoalbuminemia. Come mostrato nel nostro caso, il dosaggio della beta 2-microglobulina urinaria è sufficiente a individuare o escludere una proteinuria di basso peso molecolare.
Lapprofondimento di una qualsiasi proteinuria dovrebbe sempre includere il dosaggio della beta 2-microglobulina urinaria, al fine di individuare tubulopatie come la malattia di Dent.
Bibliografia di riferimento
Ehlayel AM, Copelovitch L. Update on Dent disease. Pediatr Clin North Am 2019;66(1):169-178. doi: 10.1016/j.pcl.2018.09.003.
Vuoi citare questo contributo?