Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Aprile 2016 - Volume XIX - numero 4
M&B Pagine Elettroniche
I Poster degli specializzandi
Una splenomegalia
per caso
Scuola di Specializzazione in Pediatria, Gastroenterologia Pediatrica, Dipartimento di Medicina Clinica e Sperimentale dell'Università di Pisa, Azienda Ospedaliera-Universitaria Pisana
Indirizzo
per corrispondenza:
giuseppe.maggiore@med.unipi.it
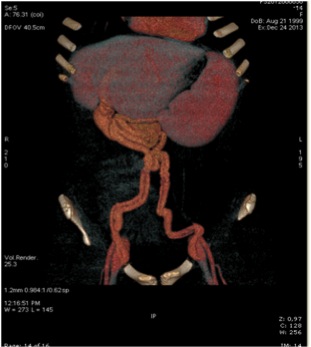
A Elettra, 14 anni e 4 mesi, viene riscontrata una epatomegalia di consistenza dura, e soprattutto una imponente splenomegalia, durante una visita in PS per un lieve incidente stradale. L'ecografia mostra la persistenza della vena ombelicale e l'ecocolordoppler la presenza di shunt porto-sistemico spontaneo tra ramo sinistro della vena porta e vena iliaca esterna, con il riscontro di rami portali intraepatici filiformi (Figura). Il suo bilancio epatico è rigorosamente normale, così come l'anamnesi familiare e personale. All'EGDS viene rilevata una gastropatia congestizia in assenza di varici esofagee.
Una RM del fegato evidenzia inoltre quattro lesioni focali con le caratteristiche della iperplasia nodulare rigenerativa. Vengono anche rilevate due formazioni litiasiche nel rene sinistro.
La biopsia epatica conferma l'ipotesi diagnostica di fibrosi epatica congenita (FEC). Un ecocardiocolordoppler con bubble test è compatibile con la presenza di piccole fistole artero-venose polmonari.
La FEC è una rara (1:20.000-1:40.000 nati vivi) ciliopatia ereditaria a trasmissione autosomica recessiva secondaria a mutazioni del gene PKHD1 che codifica la fibrocistina/poliductina. Il gene è espresso in rene, fegato, pancreas, polmone. Sono state riportate oltre 300 mutazioni, il tipo di mutazione non è predittivo dell'outcome né della patologie epatica o extra-epatica. L'epatopatia quindi si inserisce nel contesto di un disordine multiorgano a espressività variabile. LA FEC può essere isolata o associarsi tipicamente alla malattia policistica renale autosomica recessiva (ARPKD) o ad altre ciliopatie (nefronoftisi giovanile, sindrome di Meckel-Gruber, sindrome di Bardet-Biedl). Istologicamente è caratterizzata da una abnorme proliferazione duttulare con disposizione circolare al confine porto-lobulare (ductal plate malformation) con fibrosi lassa periportale ed ectasie duttali che possono interessare anche le vie biliari di medio e grande calibro (malattia di Caroli). Mentre la malattia renale, se presente, può manifestarsi già in età neonatale o nella prima infanzia, i segni e i sintomi epatici divengono manifesti nella seconda infanzia o nell'adolescenza. Le manifestazioni epatiche sono specificamente correlate da una parte allo sviluppo di ipertensione portale severa a rischio di sanguinamento e dall'altra a colangiti ricorrenti. Il bilancio bioumorale epatico è appunto tipicamente normale, ma possono essere presenti alterazioni derivanti dalle complicanze, quali: i segni di ipersplenismo (piastrinopenia e leucopenia) o le alterazioni degli indici di colestasi in caso di colangite. Non esiste una terapia specifica, essa è in genere sintomatica e diretta al trattamento delle complicanze: legatura delle varici esofagee a rischio emorragico e trattamento antibiotico per via generale in caso di colangite. In caso di ipertensione portale intrattabile può essere proposto uno shunt porto-sistemico. In rari casi la FEC sviluppa complicanze tali da diventare un'indicazione al trapianto di fegato quali: la sindrome epatopolmonare, le colangiti batteriche intrattabili o l'ascite intrattabile. Se coesiste insufficienza renale è indicato il trapianto combinato rene-fegato.
La storia di Elettra dimostra come la diagnosi possa avvenire in maniera assolutamente casuale nell'adolescenza (splenomegalia, leuco/piastrinopenia da ipersplenismo). Inoltre nel caso specifico la ripermeabilizzazione della vena ombelicale con l'imponente shunt spontaneo porto-sistemico ha fino a ora impedito la formazione di varici esofagee. Sono tuttavia osservabili quadri di iperplasia nodulare rigenerativa secondari alla anomala vascolarizzazione portale e specialmente la presenza di segni inziali suggestivi dello sviluppo della sindrome epatopolmonare, che in caso di progressione del quadro e dello sviluppo di desaturazione trasformerebbe una condizione a prognosi favorevole in una con indicazione assoluta al trapianto di organo.
Una RM del fegato evidenzia inoltre quattro lesioni focali con le caratteristiche della iperplasia nodulare rigenerativa. Vengono anche rilevate due formazioni litiasiche nel rene sinistro.
La biopsia epatica conferma l'ipotesi diagnostica di fibrosi epatica congenita (FEC). Un ecocardiocolordoppler con bubble test è compatibile con la presenza di piccole fistole artero-venose polmonari.
La FEC è una rara (1:20.000-1:40.000 nati vivi) ciliopatia ereditaria a trasmissione autosomica recessiva secondaria a mutazioni del gene PKHD1 che codifica la fibrocistina/poliductina. Il gene è espresso in rene, fegato, pancreas, polmone. Sono state riportate oltre 300 mutazioni, il tipo di mutazione non è predittivo dell'outcome né della patologie epatica o extra-epatica. L'epatopatia quindi si inserisce nel contesto di un disordine multiorgano a espressività variabile. LA FEC può essere isolata o associarsi tipicamente alla malattia policistica renale autosomica recessiva (ARPKD) o ad altre ciliopatie (nefronoftisi giovanile, sindrome di Meckel-Gruber, sindrome di Bardet-Biedl). Istologicamente è caratterizzata da una abnorme proliferazione duttulare con disposizione circolare al confine porto-lobulare (ductal plate malformation) con fibrosi lassa periportale ed ectasie duttali che possono interessare anche le vie biliari di medio e grande calibro (malattia di Caroli). Mentre la malattia renale, se presente, può manifestarsi già in età neonatale o nella prima infanzia, i segni e i sintomi epatici divengono manifesti nella seconda infanzia o nell'adolescenza. Le manifestazioni epatiche sono specificamente correlate da una parte allo sviluppo di ipertensione portale severa a rischio di sanguinamento e dall'altra a colangiti ricorrenti. Il bilancio bioumorale epatico è appunto tipicamente normale, ma possono essere presenti alterazioni derivanti dalle complicanze, quali: i segni di ipersplenismo (piastrinopenia e leucopenia) o le alterazioni degli indici di colestasi in caso di colangite. Non esiste una terapia specifica, essa è in genere sintomatica e diretta al trattamento delle complicanze: legatura delle varici esofagee a rischio emorragico e trattamento antibiotico per via generale in caso di colangite. In caso di ipertensione portale intrattabile può essere proposto uno shunt porto-sistemico. In rari casi la FEC sviluppa complicanze tali da diventare un'indicazione al trapianto di fegato quali: la sindrome epatopolmonare, le colangiti batteriche intrattabili o l'ascite intrattabile. Se coesiste insufficienza renale è indicato il trapianto combinato rene-fegato.
La storia di Elettra dimostra come la diagnosi possa avvenire in maniera assolutamente casuale nell'adolescenza (splenomegalia, leuco/piastrinopenia da ipersplenismo). Inoltre nel caso specifico la ripermeabilizzazione della vena ombelicale con l'imponente shunt spontaneo porto-sistemico ha fino a ora impedito la formazione di varici esofagee. Sono tuttavia osservabili quadri di iperplasia nodulare rigenerativa secondari alla anomala vascolarizzazione portale e specialmente la presenza di segni inziali suggestivi dello sviluppo della sindrome epatopolmonare, che in caso di progressione del quadro e dello sviluppo di desaturazione trasformerebbe una condizione a prognosi favorevole in una con indicazione assoluta al trapianto di organo.
Vuoi citare questo contributo?