Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Maggio 2015 - Volume XVIII - numero 5
M&B Pagine Elettroniche
I Poster degli specializzandi
Dura Legius sed Legius
IRCCS Materno-Infantile Burlo Garofolo, Università di Trieste
Indirizzo
per corrispondenza:
elisa.benelli@gmail.com
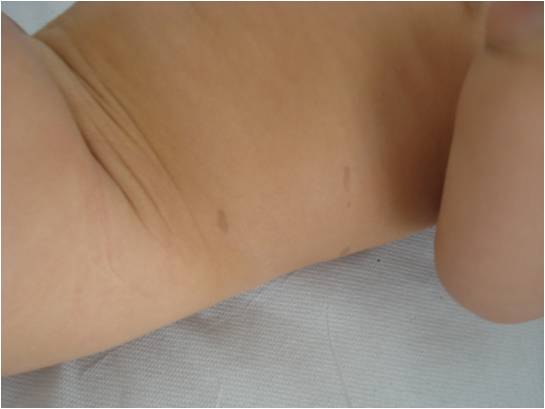
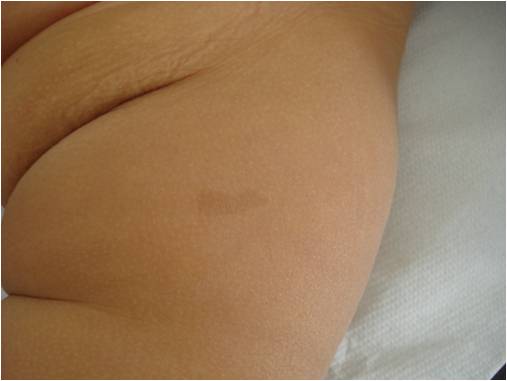
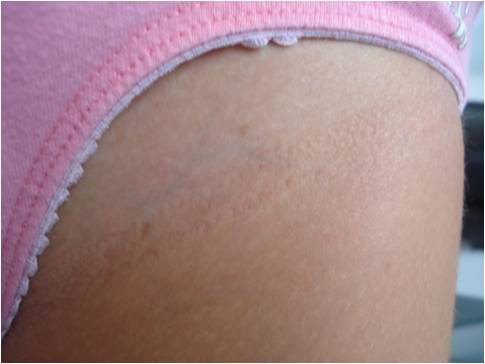
Una bambina di 8 mesi ci viene inviata dalla curante per sospetta neurofibromatosi di tipo 1 (NF1), data la comparsa, alcune settimane dopo la nascita, di numerose chiazze caffelatte (Figura 1), di cui 7 con diametro maggiore di 0,5 cm (Figura 2).
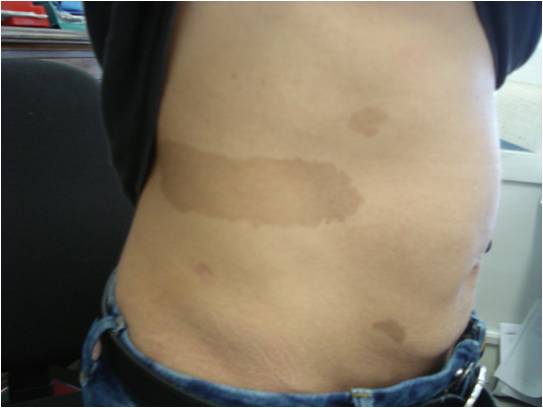
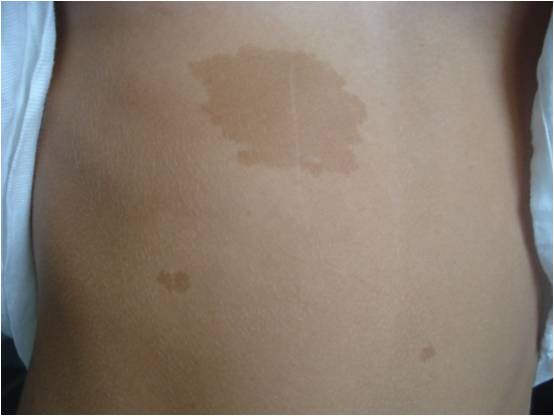
Obiettivamente non abbiamo evidenziato altri elementi tipici di NF1 (lentiggini ascellari o inguinali, noduli di Lisch, gliomi), ma ciò che ci ha colpito è stata la storia familiare in linea materna. La mamma (Figura 3), due zie (Figura 4) e il nonno materno presentano numerose chiazze caffelatte; una delle zie presenta anche lentiggini inguinali (Figura 5) e da piccola era affetta da epilessia e lieve ritardo mentale, risoltisi con le crescita.
Per questi segni la famiglia era già stata indagata e in nessun membro era stata evidenziata alcuna anomalia compatibile con NF1, ma si era riscontrata solo macrocefalia nella madre.
La storia familiare appariva suggestiva di una malattia autosomica dominante, ma una NF1 sembrava poco probabile, data la mancanza di altre manifestazioni della malattia, oltre alle chiazze.
Abbiamo quindi sospettato che si trattasse di una sindrome di Legius (SL), una forma genetica a trasmissione dominante, che si caratterizza per manifestazioni cutanee simili alla NF1 (macchie caffelatte, talvolta associate a lentiggini ascellari e inguinali), ma che non presenta le altre manifestazioni tipiche della malattia. Lanalisi genetica ha confermato la presenza della mutazione (c.973C>T) a carico del gene SPRED1, un gene regolatore del Ras/MAPK pathway.
La SL non presenta le complicanze neoplastiche e ossee tipiche della NF1 (neurofibromi, gliomi del nervo ottico, scoliosi ecc.) né i noduli di Lisch, ma può presentare macrocefalia e disturbi neuro-comportamentali (difficoltà nel linguaggio, disturbi del comportamento). Questultimo aspetto rimane dunque lunico da seguire nel follow up.
Figura 1. Chiazze caffelatte sul fianco della paziente.
Figura 2. Chiazza caffelatte con diametro maggiore di 0,5 cm.
Obiettivamente non abbiamo evidenziato altri elementi tipici di NF1 (lentiggini ascellari o inguinali, noduli di Lisch, gliomi), ma ciò che ci ha colpito è stata la storia familiare in linea materna. La mamma (Figura 3), due zie (Figura 4) e il nonno materno presentano numerose chiazze caffelatte; una delle zie presenta anche lentiggini inguinali (Figura 5) e da piccola era affetta da epilessia e lieve ritardo mentale, risoltisi con le crescita.
Figura 3. Chiazze caffelatte sul corpo della mamma della paziente.
Figura 4. Chiazze caffelatte sul corpo di una delle due zie della paziente.
Figura 5. Lentiggini inguinali di una delle due zie della paziente.
Per questi segni la famiglia era già stata indagata e in nessun membro era stata evidenziata alcuna anomalia compatibile con NF1, ma si era riscontrata solo macrocefalia nella madre.
La storia familiare appariva suggestiva di una malattia autosomica dominante, ma una NF1 sembrava poco probabile, data la mancanza di altre manifestazioni della malattia, oltre alle chiazze.
Abbiamo quindi sospettato che si trattasse di una sindrome di Legius (SL), una forma genetica a trasmissione dominante, che si caratterizza per manifestazioni cutanee simili alla NF1 (macchie caffelatte, talvolta associate a lentiggini ascellari e inguinali), ma che non presenta le altre manifestazioni tipiche della malattia. Lanalisi genetica ha confermato la presenza della mutazione (c.973C>T) a carico del gene SPRED1, un gene regolatore del Ras/MAPK pathway.
La SL non presenta le complicanze neoplastiche e ossee tipiche della NF1 (neurofibromi, gliomi del nervo ottico, scoliosi ecc.) né i noduli di Lisch, ma può presentare macrocefalia e disturbi neuro-comportamentali (difficoltà nel linguaggio, disturbi del comportamento). Questultimo aspetto rimane dunque lunico da seguire nel follow up.
Vuoi citare questo contributo?