Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Dicembre 2001 - Volume IV - numero 10
M&B Pagine Elettroniche
Pediatria per immagini
Incontinentia
Pigmenti
* Clinica
Pediatrica, IRCCS Burlo Garofolo, Trieste
°Istituto
Internazionale di Genetica e Biofisica, Napoli.
Il
caso
La
bambina, A.C., unica figlia di genitori non consanguinei
apparentemente sani, viene vista all'età di 11 mesi. Fin dalle
prime settimane di vita ha presentato una dermatite complessa,
caratterizzata da eruzione vescicolosa subentrante nella regione del
pannolino e agli arti inferiori. Le terapie prescritte per la
dermatite da pannolino e per la supposta infezione non avevano
modificato il corso della malattia che tuttavia era andata incontro
ad uno spontaneo miglioramento; già all'età di 4-5 mesi
(quando comunque clinicamente era stata sospettata la diagnosi
corretta) la fase eruttiva si era decisamente attenuata e poi del
tutto arrestata, lasciando modesti esiti pigmentari sotto forma di
chiazze vorticose iperpigmentate all'inguine e grande labbro di
sinistra (Fig 1).
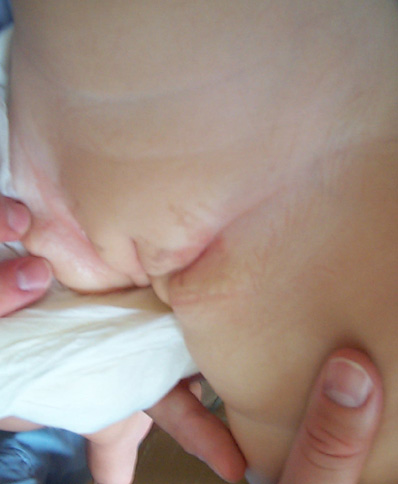
Alla
faccia posteriore della gamba destra permangono invece chiazzette
ipopigmentate disposte in maniera irregolarmente lineare (Fig 2).

La
crescita della bambina è normale, e l'esame obiettivo non
mette in evidenza altri elementi anormali, anche dal punto di vista
psicomotorio; l'oculista e (compatibilmente con l'età) lo
stomatologo non notano alterazioni.
L'analisi
molecolare mediante PCR del gene NEMO ha dimostrato la presenza di
una delezione genomica degli esoni 4-10, confermando il sospetto
clinico di incontinentia pigmenti.
L'incontinentia
pigmenti
L'Incontinentia
Pigmenti (IP; MIM 308310) è una genodermatosi X-linked
dominante caratterizzata da anomalie a carico dei tessuti originati
dalle creste neurali, soprattutto a livello della cute ove si ha una
caratteristica evoluzione attraverso quattro stadi di sviluppo.
La prima
fase, usualmente presente nelle prime settimane di vita, è
caratterizzata da una eruzione vescicolo-bollosa
infiammatoria, che può durare per alcuni mesi e che di regola
è seguita da una fase eruttiva di aspetto verrucoso. Gli
elementi sono irregolarmente distribuiti al tronco e agli arti e non
seguono una distribuzione metamerica. Alla fase verrucosa (o, in una
minoranza di casi, direttamente alla fase vescicolo-bollosa) fa
seguito la presenza di esiti pigmentari con la stessa distribuzione.
La fase pigmentaria può durare per buona parte
dell'infanzia, spesso andando incontro ad una lenta involuzione che
può comprendere la comparsa di elementi ipopigmentati con la
caratteristica distribuzione; di regola per la pubertà il
quadro cutaneo è normale.
Elementi
extracutanei sono frequenti. Nel 70% dei pazienti si osserva, infatti
una ritardata o mancata eruzione della dentizione primaria e
permanente. Il 40% dei casi presenta anomalie alle unghie delle mani
e dei piedi, nel 50% vi è alopecia. Tra i segni associati di
maggiore gravità, difetti oculari e a carico del SNC (la prima
descrizione riguardava una bambina con ritardo mentale e spasticità).
Genetica
e patogenesi: recenti scoperte
L'IP ha
una ereditarietà dominante legata al cromosoma X. La
penetranza della IP è del 100%, pur avendo un'espressività
variabile anche nell'ambito della stessa famiglia, da minime lesioni
cutanee e dentali, che vengono spesso riconosciute
retrospettivamente, fino a cospicui deficit neurologici e oculari.
Tale variabilità clinica, verificandosi anche all'interno
della medesima famiglia, non consente di prevedere il decorso della
malattia in base all'osservazione delle manifestazioni cliniche nei
parenti.
La IP si
presenta quasi esclusivamente nelle bambine (rapporto M/F = 1/37). La
mutazione risulta infatti letale per i maschi mentre le femmine
affette sopravvivono in quanto possiedono due cloni cellulari
funzionalmente diversi per effetto della lyonizzazione, un processo
mediante il quale nelle cellule somatiche si ha l'inattivazione di
uno dei due cromosomi X. Negli individui sani l'inattivazione è
casuale e avviene molto precocemente nello zigote, portando alla
presenza funzionale di entrambi i cloni cellulari; nelle femmine IP
invece si osserva un'inattivazione preferenziale del cromosoma X
mutato a favore dell'X normale. Le femmine raggiungono la maturità
sessuale e possono trasmettere il carattere alle generazioni future.
La manifestazione di IP in una figlia di donna apparentemente sana
(come nel caso presentato) può dipendere da una nuova
mutazione, oppure la madre può avere avuto manifestazioni
lievi e comunque non riconosciute.
Abitualmente
nei maschi emizigoti il fenotipo è incompatibile con la vita e
determina morte "in utero". Sono stati però
descritti in letteratura alcuni casi di maschi affetti; tale
evenienza è dovuta ad una mutazione post-zigotica, o ad una
mutazione emicromatidica in un gamete "half chromatide"
oppure a premutazioni instabili: in questo modo si realizza una
condizione definita "mosaicismo cellulare", cioè
solo alcune linee cellulari sono portatrici del difetto. Sono stati
identificati casi di maschi affetti con cariotipo 47XXY (sindrome di
Klinefelter), che probabilmente sono sopravvissuti grazie al
cromosoma X soprannumerario.
Studi di
linkage hanno assegnato il locus genico dell'Incontinentia Pigmenti
alle 2Mb distali della banda citogenetica Xq28, con un alto valore di
associazione per il gene del fattore VIII.
Recentemente,
mutazioni trovate in pazienti affetti da IP hanno permesso di
identificare la causa genetica della malattia nel difetto del gene
NF-kB essential modulator (NEMO/IKKg). (1). La proteina prodotta dal
gene NEMO é coinvolta nel pathway di attivazione dell'NF-kB,
un fattore di trascrizione ampiamente coinvolto nella risposta
immunitaria e nella resistenza agli stimoli apoptotici indotti dal
TNF. Essa infatti è una componente della proteina IKK
responsabile della fosforilazione di IKB, la proteina inibitrice che
sequestra NF-kB nel citoplasma. Di fatto nella femmina affetta da IP
non è per lo più dimostrabile alcun difetto
immunologico, dato che vengono selezionate solo le cellule
immunocompetenti che hanno attivato il cromosoma contenente l'allele
sano. Esso è invece tipicamente presente in un'altra malattia
dovuta a differenti mutazioni a carico dello stesso gene NEMO, che si
esprime solo nel maschio con immunodeficienza, elevati valori di IgM
e displasia ectodermica ipoidrotica (2).
Analizzando
357 pazienti IP, tra casi familiari e sporadici, in collaborazione
con l'International IP Consortium (IPIF), è stato dimostrato
(3) che nell' 83% dei casi, è presente una delezione tra
l'esone 4 e l'esone 10, dovuta ad una ricombinazione sbilanciata tra
sequenze ripetute (LTR) presenti all'interno dell'introne 3 di NEMO e
fuori dell'esone 10. Il risultato in questo caso sarà di
un'anomala informazione alle strutture che, ricevuto il messaggio,
devono provvedere a formare il prodotto del gene e quindi viene
impedita la funzione biologica della proteina. Sono state inoltre
evidenziate sostituzioni nonsenso e missenso, di cui la maggior parte
concentrate nella parte finale del gene, essenziale per la stabilità
e la funzione stessa della proteina.
Sono
stati eseguiti, inoltre, dei saggi di funzionalità del gene
nei pazienti mutati e si é visto che l'attivazione di NF-kB é
difettiva nelle cellule IP. Questa è la prima evidenza di una
malattia mendeliana associata ad un difetto nell'attivazione
dell'NF-kB e per questo fornirà nuove conoscenze sul ruolo di
questo pathway nell'uomo.
L'identificazione
del gene responsabile dell'IP rende possibile la diagnosi molecolare
della malattia, particolarmente importante per quanto concerne le
applicazioni immediate, quali la diagnosi presintomatica anche a
livello prenatale. E' stato possibile infatti mettere a punto un
rapido test molecolare per la diagnosi della malattia, che mette in
evidenza la presenza del riarrangiamento disponendo di una piccola
quantità di DNA estratto da un comune prelievo di sangue o dai
villi coriali. Nei soggetti che non presentano la delezione la
diagnosi molecolare è resa complessa dalla presenza di uno
pseudogene a circa 75 kb dal gene stesso, che conserva il 99,9 % di
identità (4). Per questo motivo sono state utilizzate
strategie molecolari più sensibili come DHPLC e sequenziamento
diretto per un'analisi di mutazione gene specifica.
Bibliografia
1. Smahi
A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, Israel A,
Heiss NS, Klauck SM, Kioschis P, Wiemann S, Poustka A, Esposito T,
Bardaro T, Gianfrancesco F, Ciccodicola A, D'Urso M, Woffendin H,
Jakins T, Donnai D, Stewart H, Kenwrick SJ, Aradhya S, Yamagata T,
Levy M, Lewis RA, Nelson DL. "Genomic rearrangement in NEMO
impairs NF-kappaB activation and is a cause of incontinentia
pigmenti. The International Incontinentia Pigmenti (IP) Consortium."
Nature. 2000 May 25;405(6785):466-72.
2. A
Jain, CA Ma, S Liu, M Brown, J Cohen, W Strober.Specific missense
mutations in NEMO
result in
hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat
Immunol 2001;2:223-8
3.
Aradhya S., Woffendin H., Jakins T., Bardaro T., Esposito T., Smahi
A., Shaw C., Levy M., Munnich A., D'Urso M., Lewis R.A., Kenwrick S.
and Nelson D.L.. (2001)"A recurrent deletion in the ubiquitously
expressed NEMO (IKKgamma) gene accounts for the vast majority of
Incontinentia Pigmenti mutations" Human Molecular Genetics ,
(10)19; 2171-2179
4 Aradhya
S., Bardaro T., Yamagata T., Galgoczy P., Esposito T., Patlan H.,
Ciccodicola A., Kenwrick S., Platzer M., D'Urso M. and Nelson D.L..
(2001). "Multiple pathogenetic and benign genomic rearrangement
occur at a 35-kb duplication involving the NEMO and LAGE2 genes".
Hum Mol Genet. (10)22:2557-2567
Vuoi citare questo contributo?