Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Ottobre 2001 - Volume IV - numero 8
M&B Pagine Elettroniche
Pediatria per immagini
Istiocitosi
cefalica benigna
La
bambina si è presentata al medico curante all'età di 16
mesi per la presenza di numerose e grosse papule al viso comparse
venti giorni prima e considerate come grossi foruncoli. Questi
elementi hanno avuto una eruzione pressochè contemporanea nel
giro di due tre giorni e hanno subito assunto l'aspetto che si vede
nella foto scattata dai genitori stessi. ( Fig 1).
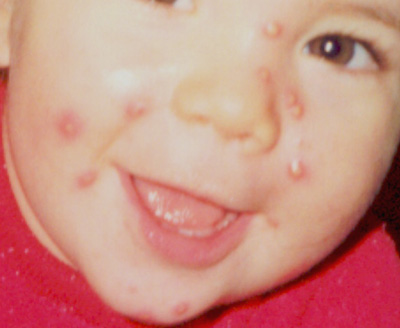
Si tratta
di grosse papule di alcuni mm di diametro color carne disposte alla
zona centrale del volto, cupoliformi di consistenza parenchimatosa
alcune delle quali delimitate da un franco alone eritematoso e del
tutto asintomatiche. Il restante esame obiettivo generale era
negativo.Nella storia clinica precedente non vengono segnalate
lesioni cutanee né altre patologie. La terapia prescritta dal
curante, una crema antibiotico-cortisonica (Gentalyn beta) e un
antibiotico per via generale (Cefaclor) praticata per una decina di
giorni non ha portato a variazioni significative (Fig 2) anche se due
elementi alla sinistra della piramide nasale mostravano l'evidenza di
una lieve ombelicatura centrale in precedenza non visibile.
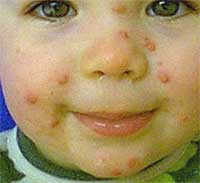
La
diagnosi appariva incerta: La morfologia inziale della dermatite era
caratterizzata da una espressione "florida"sia per
l'aspetto molto rilevato delle papule (quasi emisferiche) che per
l'eritema alla base; per questo motivo, oltre che per la ombelicatura
centrale evidenziata in due elementi, si è pensato anche ad un
mollusco contagioso; tuttavia le papule di questa virosi hanno un
tipica tonalità perlacea e traslucida ed un materiale
lattesecnte può apparire a livello della ombelicatura
centrale; tutti elementi qui totalmente mancanti. La assenza di una
sfumatura giallo-arancio non fa pensare alla dermatite papulonodulare
più facilmente identificabile nei primi anni di vita e cioè
allo Xantogranuloma Giovanile (JXG). L'aspetto così esuberante
invece non ha fatto riconoscere immediatamente una tipica se pur rara
patologia che colpisce elettivamente il viso del bambino nei primi
anni di vita e cioè la Isticitosi Cefalica Benigna (BCH) che
si manifesta appunto elettivamente al volto con una eruzione di
maculopapule giallastro-roseo-rossso brunastre; in questo nostro caso
l'aspetto francamente cupoliforme delle lesioni e la presenza di un
alone infiammatorio sono stati elementi confondenti. Inoltre la
presenza della dermatite solo in una zona delicata come il viso ci ha
trattenuto dall'essere incisivi e tempestivi nelle indagini
". Fortunatamente " però dopo alcune
settimane è comparsa una lesione simile alle precedenti al
gomito destro. Viene eseguita perciò in quella sede biopsia
cutanea. All'esame istologico si evidenzia la presenza di una flogosi
granulomatosa con un infiltrato cellulare nella parte alta e media
del derma composto prevalentemente da istiociti multinucleati, che
alla immunoistochimica avevano caratteristiche di istiociti
(positività per l'antigene CD68 -KP1) ma non di cellule di
Langerhans (negatività per l'antigene CD1a -OKT6- e per la
proteina S100); non si sono inoltre individuati depositi di lipidi
all'interno degli istiociti, come è evidente nella fase
florida dello JXG.
Viene
pertanto confermata la diagnosi di BCH. Spieghiamo ai genitori la
benignità della patologia e la evoluzione verso regressione
spontanea in un tempo non breve (alcuni anni) e la non necessità
di terapia.
Quattro
mesi dopo la bambina è stata rivista per una febbricola di
breve durata. In questa occasione le lesioni avevano perso il loro
aspetto "tumultuoso" ed apparivano più facilmente
inquadrabili nella diagnosi di BCH. (Fig 3)
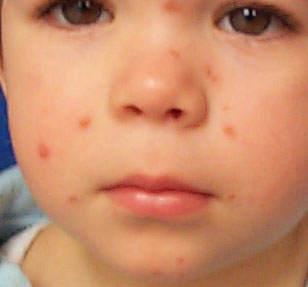
Le papule
erano più piatte, con un colorito rame - bruniccio ed
apparivano solo lievemente ridotte rispetto al controllo precedente;
sono rimaste sempre del tutto asintomatiche; anche in questa
occasione il restante esame obiettivo era negativo.
E' stato
il prof F. Gianotti che per primo nel 1971 ha descritto una eruzione
cutanea autorisolutiva che interessa prevalentemente il volto del
bambino dei primi anni di vita i cui elementi costitutivi sono
maculo- papule piatte o cupoliformi di colorito variabile (carneo
-giallastro-bruno rossiccio ) che non colpisce mai le mucose e gli
organi interni e non compromette lo stato di salute; le papule
risultano composte di istiociti che non hanno le caratteristiche
delle cellule di Langerhans. L'eruzione interessa tipicamente i primi
tre anni di vita e la evoluzione è sempre favorevole in
pressochè tutti i casi fino ad ora descritti con scomparsa
delle lesioni in un tempo non brevissimo ( una media di cinque- sei
anni) e la possibilità di qualche esito in macule brune o
atrofiche. La denominazione di Istiocitosi Cefalica Benigna è
pertanto ancora molto appropiata per descrivere questo tipo di
patologia del volto del bambino nei primi anni di vita.
Le
caratteristiche degli istiociti presenti nelle lesioni sono peculiari
e permettono una chiara distinzione dalle isticitosi a cellule di
Langerhans (Istiocitosi di classe I): non hanno infatti il
caratteristico nucleo reniforme ma un nucleo pleomorfo con citoplasma
abbondante eosinofilico e pallido e non infiltrano mai l'epidermide
come è invece evidente nelle isticitosi di classe I. Anche al
microscopio elettronico la morfologia è caratterizzante e
diversa da quella tipica delle isticitosi a cellule di Langerhans:
non si dimostra la presenza dei granuli di Birbek ma gli elementi
distintivi che si rinvengono in una buona parte di queste cellule
sono organelli a forma di virgola o di S o vermiformi e vescicole "
laminate" Alla immunoistochimica gli istiociti reagiscono con
anticorpi monoclonali diversi rispetto alle isticitosi- Langerhans:
sono infatti negativi per anticorpi CD1 e per la proteina S100 mentre
sono CD68 positivi.
Sono
conosciute altre istiocitosi "benigne" autorisolutive a
cellule non Langerhans (Istiocitosi cutanee di classe II ) tutte
abbastanza simili tra di loro. Come alla BCH , anche ad ognuna di
esse sono state attribuite alcune caratteristiche cliniche ed
istologiche peculiari.
Lo JXG
(la più frequente e già sopra descritta ) ha come
substrato istologico suggestivo (solo però nella fase
avanzata) la presenza di istiociti con citoplasma infarcito di lipidi
( responsabili della colorazione giallo arancio delle lesioni) con
aspetto "schiumoso"che in parte , nelle lesioni mature, si
trasformano in cellule giganti multinucleate (cellule di Touton).
L'Istiocitosi
Generalizzata Eruttiva (GEH) ha la stessa istologia della BCH ,
la stessa lenta e favorevole evoluzione spontanea, si manifesta a
gettate con numerosissime piccole papule giallastre o bruno-roeso su
tronco viso ed arti ma è molto rara nei bambini. Altrettanto
raro è lo Xantoma Disseminato (XD) che ha più o
meno la stessa manifestazione clinica della GEH con numerossimi
elementi papulosi che possono infiltrare anche labbra congiuntive
faringe e laringe: gli istiociti in questo caso possono essere
infarciti di lipidi ed avere l'aspetto delle cellule schiumose. In
conclusione tutte queste istiocitosi non Langerhans sono strettamente
correlate fra loro, hanno aspetti con ampio margine di
sovrapposiszione e vengono oggi considerate come espressione di
diverse fasi dello stesso dello stesso disordine istiocitario
piuttosto che entità nettamente separate.
La BCH
può rappresentare una forma localizzata di GEH e o anche una
fase abortiva di uno JXG A sostegno di ciò sta anche il fatto
che sono descritti due casi di BCH evoluti in JXG : in uno di questi
tale progressione si è realizzata a un anno di distanza dalla
sua comparsa e dopo una infezione virale (Varicella) .Non vi sono
invece, per questo gruppo di malattie cutanee,segnalazioni di
trasformazione verso una proliferazione maligna. E' stato però
di recente riportato il singolo caso di una bambina che ha sviluppato
un diabete insipido a un anno di distanza da una eruzione
maculopapulosa al volto clinicamente e istologicamente inquadrata
nella diagnosi di BCH. Fino ad ora il diabete insipido è stato
associato solo allo XD oltre che, come noto e più
frequentemente ,alle istiocitosi a cellule di Langerhans.
Bibliografia
Benign
cephalic histiocytosis Gainotti F, Caputo R , Ermacora E, Gianni E
Arch Dermatol 1986 Sep; 122 (9): 1038-43
Non-
Langerhans cell histiocytosis. A new unifying concept. Zelger BW,
Sidoroff A, Orchard G, Cerio R Am J Dermatopathol 1996 Oct; 18(5):
490- 504
Benign
cephalic histhiocytosis with diabetes insipidus Rodriguez Wl, Travers
SH, et al Pediatr Dermatol 2000 Jul-Aug ; 17(4): 296-8
Benign
cephalic histhiocytosis progressing into juvenile xanthogranuloma: a
non -Langerhans cell histiocytosis tranforming under the influence of
a virus? Rodriguez-JuradoR, Duran-McKinsterC,Ruiz_Maldonado R. Am J
Dermatopathol 2000 Feb;22(1):70-4
Vuoi citare questo contributo?