Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Marzo 2009 - Volume XII - numero 3
M&B Pagine Elettroniche
Pediatria per l'ospedale
Neurofibromatosi
tipo I (parte seconda)
Membro
della Commissione Nazionale Vaccini
Indirizzo
per corrispondenza: bartolozzi@unifi.it
Gliomi
delle vie ottiche
I gliomi
delle vie ottiche, tipicamente astrocitomi pilocitici di basso
grado, sono i più comuni tipi di tumori maligni endocranici
nei pazienti con neurofibromatosi tipo I. I glomi, associati alla
neurofibromatosi tipo I (NF1), sono localizzati più spesso
lungo il nervo ottico (vedi D nella figura 2), mentre i gliomi
sporadici si ritrovano più spesso al chiasma o dopo il
chiasma.
I gliomi
del nervo ottico si ritrovano in circa il 15% dei bambini con NF1 e
usualmente compaiono nella prima decade di vita. Essi hanno un
decorso benigno e solo da un terzo alla metà dei pazienti con
gliomi del nervo ottico, associati alla NF1, manifesta dei sintomi. I
soggetti con sintomi presentano proptosi, perdita della vista e
pubertà precoce per sofferenza ipotalamica. Confrontando i
sintomi dei gliomi sporadici con quelli legati alla NF1, risulta che
questi ultimi presentano più spesso pubertà precoce,
mentre i segni di aumentata pressione intracranica sono meno comuni.
La
pubertà precoce si vede nel 12-40% dei bambini con
tumori del chiasma: il suo segno più precoce è
l'aumento improvviso della velocità di crescita staturale.
Poiché i bambini non si lamentano di disturbi della visione, è
necessario prevedere una visita oftalmologica ogni anno nei
bambini al di sotto dei 6 anni, anche perché questi bambini
sono quelli che più spesso sviluppano un glioma del nervo
ottico. Una risonanza magnetica per diagnosticare i gliomi
dell'ottico asintomatici non è giustificata: una visita
annuale da un neuro-oftalmologo è il protocollo usato per
lesioni che si presentano senza sintomi.
La
progressione della lesione è meno comune di quello che si
osserva nei gliomi sporadici: il trattamento va preso in
considerazione soltanto nei pazienti che hanno sintomi e segni
oculari ed endocrinologici. In caso di gliomi dell'ottico
aggressivi, associati alla NF1, è necessario fare una
risonanza magnetica di base, per valutare la velocità di
crescita, e la chemioterapia con carboplatino e vincristina.
La terapia radiante, sebbene sia efficace, è scoraggiante nei
bambini con NF1 per l'aumentato rischio di un secondo tumore e di
stenosi vascolari.
I gliomi
possono avvenire anche a carico del tronco cerebrale, del diencefalo
e del cervelletto in più del 3,5% dei pazienti con NF1.
Confrontando i gliomi del tronco della popolazione in generale con
quelli legati alla NF1, si risconta un decorso più spesso
senza dolore e una regressione spontanea. Essi si possono manifestare
talvolta come astrocitomi di alto grado, per cui viene consigliata
una risonanza magnetica per riconoscere la velocità di
crescita per i tumori gliali non-ottici, aggressivi e per masse
tumore-simili nei pazienti con NF1. Il trattamento chirurgico non è
raccomandato a meno che la lesione non presenti una crescita rapida o
si assista a un deterioramento dello stato del pazienti. Come per i
gliomi dell'ottico, è la localizzazione del tumore che
indica la necessità di un trattamento chirurgico
individualizzato.
Alterazioni
cardio-vascolari
Le
manifestazioni cardio-vascolari della NF1 includono le malattie
congenite di cuore, le vasculopatie e l'ipertensione.
Le
coronaropatie avvengono nella NF1 più spesso di quanto non si
ritrovi nella popolazione generale. La stenosi dell'arteria
polmonare rappresenta il 25% delle malformazioni cardiache. Tutti i
bambini debbono essere sottoposti all'ascoltazione del cuore e alla
misurazione della pressione arteriosa. Ogni soffio deve essere
valutato da un cardiologo ed esaminato con l'ecocardiografo.
Le
vasculopatie nella NF1 sono rappresentate da stenosi, aneurismi e
malformazioni artero-venose, che rappresentano la seconda causa di
morte in questi soggetti. Le vasculopatie di regola interessano il
sistema arterioso e la stenosi dell'arteria renale è la
manifestazione più comune, presente nell'1% dei pazienti con
NF1. L'arteriografia renale è indicata in ogni paziente con
ipertensione e NF1. Malattie cerebrovascolari, specialmente nei
pazienti più giovani, sono il risultato di stenosi e
occlusioni e sono diagnosticate in bambini con un quesito clinico di
stanchezza, movimenti involontari, cefalea o convulsioni secondarie a
ischemia. Fisiologicamente le lesioni vascolari mostrano displasia
fibromuscolare con ispessimento dell'intima e proliferazione delle
cellule di Schwann, senza arteriosclerosi.
L'ipertensione
è significativamente associata alla NF1: la pressione
arteriosa va controllata annualmente con un limite < 140/90 mm Hg.
La stenosi dell'arteria renale è la situazione più
frequente. Tuttavia possano essere riscontrati la coartazione
dell'aorta e il feocromocitoma. Il feocromocitoma si ritrova con
una frequenza dallo 0,1 al 5,7%. La maggior parte (90%) sono benigni
e tipicamente avvengono nella popolazione adulta. Tuttavia per il
rischio di malignità, in ogni paziente con ipertensione,
specialmente se di tipo parossistico, o con sintomi di eccesso di
catecolamine, come la cefalea, la sudorazione, le palpitazioni o
l'ansietà, debbono essere misurate nelle 24 ore la
concentrazione urinaria di catecolamine totali e frazionate e i loro
metabolici. Soltanto dopo che la presenza di un feocromocitoma sia
stata confermata biologicamente, va eseguita la RM per localizzare il
tumore. C'è un'associazione fra feocromocitoma e tumori
carcinoidi, usualmente nel duodeno, per cui la presenza di uno deve
indurre il clinico a ricercare l'altro.
Deficit
neuro-cognitivi
I decifit
neurocognitivi sono le complicazioni più spesso riportate
nella NF1. I deficit dell'apprendimento nei bambini con NF1 possono
includere deficit viso-spaziali e viso-motori e disordini del
linguaggio. In aggiunta ai deficit specifici, non verbali e verbali,
visti nel 30-60% dei bambini con NF1, sono comuni i deficit della
coordinazione motoria, fini e grossolani.
Nel
fenotipo cognitivo della NF1, hanno la maggiore incidenza il deficit
di attenzione/iperattività (ADHD), disordini dello spettro
autistico, comportamenti anormali e comportamenti psico-sociali. I
bambini con tipico ADHD rispondono bene al metilfenidato (Ritalin).
Anche la terapia cognitiva comportamentale può essere utile. I
pazienti con NF1 usualmente dimostrano livelli di QI nel gruppo
medio-basso; anche la frequenza del ritardo mentale è
aumentata. E' necessaria una valutazione ogni anno.
Alla
risonanza magnetica i bambini con NF1 hanno regioni iperintense alle
sequenze T2, qualche volta chiamate oggetti brillanti non
identificati (UBO). Questi UBO a volte sono difficilmente
distinguibili da gliomi di bassa crescita.
Genetica
e prove genetiche
Tutti i
soggetti con NF1 sono nati con una copia funzionante e una non
funzionante del gene NF1 in ogni cellula del loro corpo. Circa la
metà di tutti i casi non presenta una storia familiare e va
quindi considerata come una nuova mutazione. Circa l'80% nelle
mutazioni non mendeliane del gene NF1, insorte come nuove, sono di
origine genitoriale. Un aspetto essenziale delle manifestazioni
cliniche è la loro espressione variabile, che rende difficile
esprimere dei pareri nei confronti dei consigli e della prognosi. La
condizione deriva da una mutazione della linea germinale del gene NF1
localizzato sul cromosoma 17q11.2.
Il
mosaico NF1, una variante della NF1, inizialmente chiamata
neurofibromatosi segmentale da Riccardi ed Eichner, risulta da
una precoce mutazione somatica e può presentarsi con un'ampia
varietà di aspetti clinici. A seconda del grado di mosaicismo,
si riconoscono 3 forme:
- Malattia generalizzata lieve
- Malattia localizzata
- Mosaicismo unicamente gonadaleMalattia generalizzata lieve
I
soggetti con mosaicismo sono a minor rischio di presentare gravi
complicazioni mediche.
Recentemente
mutazioni nel gene SPREDI sono state trovate in molte famiglie,
segreganti macchie autosomico dominanti caffè e latte.
Circa il
5% dei pazienti con NF1 ha una delezione intera o quasi intera del
gene NF1.
Questi
pazienti mostrano un fenotipo più grave, incluso l'inizio
più precoce, un maggior numero di neurofibromi, aspetti
dismorfici del volto, aumentato rischio di malignità e
interessamento del tessuto connettivo, come lassità
articolare, cute iperestensibile e prolasso della valvola mitrale.
L'ecocardiografia va eseguita per identificare il prolasso della
mitrale in soggetti con microdelezioni del gene IF1.
Le prove
genetiche nella NF1 sono complesse per il gran numero di possibili
mutazioni in un grande gene. L'analisi dell'ereditarietà
può essere eseguita, ma non è utile per il numero
elevato di soggetti sporadicamente colpiti
Neurofibromina
(proteina prodotta dal gene NF1)
La
proteina formata dal gene NF1 (neurofibromina) è una proteina
citoplasmatica, lunga 2.818 aminoacidi. Poiché i pazienti con
NF1 sono soggetti a sviluppare tumori benigni e maligni, si pensa che
la neurofibromina abbia una funzione di soppressione dei tumori e sia
un regolatore di crescita negativo. La neurofibromina limita la
crescita cellulare e la sua assenza o la sua ridotta espressione
portano a un aumento della crescita cellulare. E' probabile che
essa funzioni come un regolatore negativo del Ras, una proteina
intracellulare di segnalazione, importante per la regolazione della
crescita e della sopravvivenza delle cellule. La neurofibromina
inibisce l'attività delle proteine GTPasi del Ras,
catalizzando l'idrolisi del Ras attivo, legato al
guanosin-trifosfato, fino al passaggio a Ras inattivo legato al
guanosin-difosfato (vedi Figura 4). La
perdita della neurofibromina porta a un'attività non
controllata del Ras e costituisce un segnale di arresto e quindi di
aumento della crescita cellulare. La deregolazione dell'attività
del Ras porta all'attivazione di molti importanti intermediari a
valle, inclusa la proteina rapamicina dei mammiferi (mTor). Oltre
alla regolazione del Ras la neurofibromina funziona anche nei
confronti della regolazione positiva dei livelli di adenosin
monofosfato ciclico (AMP). L'aumento dei livelli di AMP ciclico si
associa a ridotta crescita cellulare, attraverso l'interferenza di
molte vie mitogeniche di segnalazione.
Nuove
terapie
Le nuove
terapie per i tumori, associati alla NF1 possono essere raggruppate
in:
- Quelle che mirano a deregolare le vie di segnalazione all'interno delle cellule tumorali e
- Quelle che mirano a modificare la componente stromale nel microambiente del tumore.
Inoltre
sono state proposte strategie basate sulla correzione dei segnali del
Ras per il trattamento dei deficit cognitivi dei bambini con NF1.
Nei
confronti delle cellule tumorali
Poiché
la neurofibromina funziona come un inibitore del Ras, i primi studi
si sono concentrati sugli inibitori del Ras. Il tipifarnib è
un inibitore della protein transferasi farnesyl, che inibisce la
farnesilazione e la geranilgeranilazione del Ras, richieste per il
suo trasferimento alla membrana cellulare e alla successiva
attivazione. Sono state di recente completate prove in fase 1 con il
tipifarnib, eseguite nei bambini con tumori solidi refrattari o con
neurofibromi plessiformi in corso di NF1; il farmaco è stato
ben tollerato nei bambini e negli adulti. E' in corso una prova in
fase II con il tipifarnib in pazienti con neurofibroma plessiforme
associato alla NF1.
Figura
4. La NF1 dipende dall'aberrante funzione della neurofibromina
e dalla disregolazione di molte vie di segnalazione di numerosi tipi
di cellule.
GF
= fattore di crescita; GPCR = recettore della proteina G; GFR =
recettore del fattore di crescita; tutti gli altri acronimi dei
componenti della cascata di segnalazione sono usati secondo le solite
indicazioni.
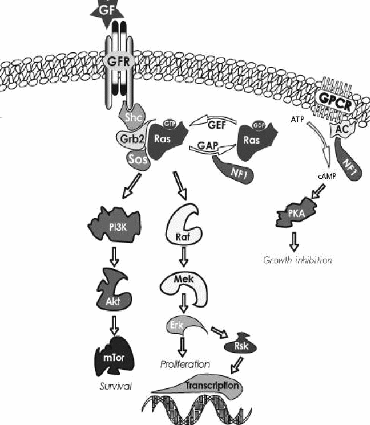
Poiché
la neurofibromina regola le segnalazioni di mTor, l'uso della
rapamicina e dei suoi analoghi va considerato nel trattamento dei
tumori in soggetti con NF1. La rapamicina fu inizialmente descritta
come un farmaco immunosoppressivo che si lega al suo obiettivo
FKBP12, un inibitore delle segnalazioni di mTor. Interesse per l'uso
della rapamicina è stato sollevato recentemente dalla
descrizione che la rapamicina per bocca determina una regressione
degli astrocitomi giganto-cellulari subependimali, in un piccolo
numero di pazienti con sclerosi tuberosa.
Nei
confronti della componente stromale
Agenti
antistaminici, come il chetotifene, pur non essendo dotati di
alcun effetto favorevole per il trattamento dei neurofibromi
plessiformi, sono usati perché porterebbero a un'attenuazione
dei sintomi soggettivi.
I
neurofibromi plessiformi mantengono un'abbondante
vascolarizzazione, suggerendo che agenti terapeutici, che agiscono
sulla vascolarizzazione del tumore, possono essere efficaci.
Venne
usato per primo l'interferon α, ma l'applicazione pratica fu
deludente. L'AZD2171, che inibisce il recettore della
tirosin-chinasi è conosciuto come inibitore dell'angiogenesi;
questa sostanza e un altro inibitore dell'angiogenesi (la
talidomide) sono probabilmente efficaci nel trattamento dei
tumori della guaina dei nervi periferici. L'AZD2171 è usato
in fase 1 in pazienti con neurofibromi plessiformi e neurofibromi
spinali.
Il
pirfenidone, una sostanza antifibrotica che aiuta a ridurre le
attività delle citochine, liberate dai fibroblasti in
vicinanza dei neurofibromi, rende incapace di agire la rete cellulare
di supporto (fibroblasti, mast cellule e altri). E' stata
completata di recente una prova in fase II con pirfenidone per bocca
in 24 pazienti con NF1: 4 mostrarono una regressione del tumore, 3
una progressione e in 17 la malattia si stabilizzò. Questo
farmaco è in ulteriore valutazione in pazienti pediatrici.
Nuovi
trattamenti per i disturbi dell'apprendimento
Recenti
studi nel topo con nf+l- sono in corso con la lovastatina. Gli studi
hanno mostrato un comportamento spaziale e di apprendimento normale
neri topi trattati, mentre nei topi trattati con placebo o non
trattati erano presenti alterazioni del comportamento.
Future
direzioni
Grazie
alla destinazione di più di 200 milioni di $, da parte
Programma per la Neurofibromatosi, sono stati ottenuti molti
progressi. Sono previste in un prossimo futuro prove in fase II per
l'uso della rapamicina e del sorafenib nei tumori plessiformi e
della lovastatina nei disturbi dell'apprendimento.
Conclusioni
L'attuale
trattamento della NF1 si basa sui consigli genetici e sul trattamento
sintomatico di specifiche complicazioni. Accanto alle prove cliniche
si sono dimostrate molto utili le ricerche nei piccoli animali di
laboratorio, come i topi, con situazioni patologiche simili a quelle
umane.
Considerazioni
personali
La
rivista sulla neurofibromatosi tipo 1, sopra riportata, dopo quella
famosa di Riccardi, comparsa sul N Engl J Med (Riccardi VM, Von
Recklinghausen neurofibromatosis. N Engl J Med 1981;305:1617-27),
rappresenta un punto fermo, molto aggiornato, al quale far
riferimento quando si parli di questa malattia. Essa è
completa sotto ogni riguardo, dagli aspetti clinici agli aspetti
genetici, biochimici, patogenetici e ai tentativi terapeutici.
Non si
interessa tuttavia delle difficoltà diagnostiche e dei
comportamenti pratici che il pediatra deve affrontare quando si trovi
davanti a un bambino di pochi anni, con qualche macchia caffè
e latte, senza nessun altro sintomo di neurofibromatosi e con una
storia familiare assolutamente negativa: al curante, di fronte alle
macchie caffè e latte viene un dubbio, al quale non è
in grado di rispondere durante l'esame obiettivo e che dovrebbe
assolutamente tenere dentro di sé, senza esternarlo ai
genitori. In assenza di esami di laboratorio semplici, essendo ancora
troppo presto per cercare, presso un oculista pediatra, preparato ad
affrontare problemi del genere, i noduli di Lisch che si presentano
fra i 5 e i 10 anni, avendo a che fare con una malattia non
trattabile, anche quando riconosciuta precocemente, il pediatra
dovrebbe solo scrivere sulla cartellina del bambino la presenza delle
macchie caffè e latte, senza commento, per ricordare, a ogni
visita successiva, la necessità d'impostare ogni volta
un'accurata diagnosi.
Risparmiare
ai genitori anni di apprensioni e di ansie deve essere un obiettivo
da perseguire. Mi è capitato più volte di dover
visitare un piccolo bambino, giunto a visita per confermare o negare
una diagnosi o un sospetto diagnostico di neurofibromatosi,
incautamente offerto ai genitori. La sensazione d'impotenza e
d'incapacità di dare una risposta definitiva, ancora mi
attanaglia. Oggi, attraverso internet, i genitori si rendono
immediatamente conto della gravità della diagnosi, della
mancanza di cure e della incertezza della prognosi, legata anche
all'ampiezza del ventaglio del quadro clinico e dell'evoluzione
della malattia. La convivenza con una diagnosi incerta di una
malattia non curabile rappresenta per i genitori e anche per il
pediatra di famiglia, una situazione ai limiti della sostenibilità.
Raggiunta
un'età adeguata, oltre gli 8-10 anni, la negatività
del quadro clinico, al di là delle macchie caffè e
latte, farà rientrare il sospetto o, come purtroppo può
accadere in qualche raro caso, la comparsa di un neurofibroma o
l'evidenza, anche alla semplice ispezione, di un possibile nodulo
di Lisch, deve far scattare la necessità di sottoporre il
bambino a esami specialistici, per i quali è ovviamente
richiesta la partecipazione cosciente dei genitori. Anche in
questo caso deve sempre prevalere da parte del curante un
atteggiamento di sostegno psicologico ai genitori, per accompagnarli
nel difficile iter diagnostico e nei burrascosi anni a seguire.
Avere a
che fare con una situazione del genere rappresenta una prova decisiva
per il pediatra, nella quale, una volta di più, egli deve
usare tutte le sue doti di umanità e di preparazione.
Vuoi citare questo contributo?