Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Settembre 2011 - Volume XIV - numero 7
M&B Pagine Elettroniche
Le Giornate di Medico e Bambino
Medico
e Bambino 2011
Mestre (Venezia),
6-7 maggio 2011
Pubblichiamo
qui di seguito gli abstract dei giovani del congresso Le
Giornate di Medico e Bambino
Sessione
orale
Sessione
poster
Infezione
post-natale da CMV come trigger di grave espressione clinica di BRIC
tipo 2 in eterozigosi
SESSIONE
ORALE
Dipartimento
Materno-Infantile, Azienda Ospedaliero-Universitaria Pisana
Scuola di Specialità in Pediatria, Università di Pisa
Scuola di Specialità in Pediatria, Università di Pisa
La
malattia di Wilson è una malattia autosomica recessiva dovuta
a mutazioni del gene ATP7B, che codifica per la principale proteina
coinvolta nel metabolismo del rame. In età pediatrica la
malattia si manifesta quasi esclusivamente con un interessamento
epatico eventualmente sintomatico, mentre l'esordio dei sintomi
neurologici è più tardivo e interessa il giovane
adulto. Il trattamento con d-penicillamina è generalmente
efficace e ben tollerato.
Riportiamo
il caso di un adolescente caucasico di 17 anni presentatosi alla
nostra attenzione per la comparsa da circa un anno di difficoltà
di concentrazione, facile affaticabilità e astenia
ingravescente, e più recentemente comparsa di disfagia,
disartria, e difficoltà nella deambulazione autonoma. L'esame
neurologico denotava ipostenia ai quattro arti, scarsa motilità
attiva, lieve aumento del tono di fondo, con atteggiamento distonico,
tremori fini alle estremità e fascicolazioni della lingua.
L'elettroencefalogramma mostrava modico rallentamento delle
frequenze di base. La RM metteva in evidenza alterazioni simmetriche,
dei nuclei della base. Il sospetto diagnostico di M. di Wilson è
stato confermato dalla presenza di una ceruloplasmina indosabile e di
una cupruria di 100 ?g/24 ore. Inoltre l'esame oftalmologico non
evidenziava l'anello di Kayser-Fleischer, clinicamente non c'era
epato- e/o splenomegalia e il bilancio epatico bioumorale era
normale. Ciononostante esistevano segni ecografici suggestivi di
epatopatia cronica con evidenza di ipertensione portale e una
elastometria epatica suggestiva di cirrosi, poi confermata all'esame
istologico. La ricerca di mutazioni del gene ATP7B mostrava una
mutazione monoallelica 2532delA nell'esone 10.
Il
paziente iniziava il trattamento con dosi crescenti di
d-penicillamina fino a una dose di 300 mg raggiunta dopo 3 settimane,
ma si assisteva alla improvvisa comparsa di clonie agli arti e di un
importante peggioramento dell'atteggiamento distonico già
presente. Nell'ipotesi che il peggioramento dei sintomi neurologici
fosse stato scatenato dalla d-penicillamina si sostituiva il
trattamento con solfato di Zn con lento beneficio.
Questo
caso presenta alcune peculiarità quali l'esordio neurologico
apparentemente puro inusuale in età pediatrica, la
presenza di una grave epatopatia cirrotica silente sia da un punto di
vista clinico che di laboratorio e infine l'aggravamento dei
sintomi neurologici correlabile con la somministrazione di
d-penicillamina. Questa complicanza, nota nell'adulto, può
interessare dal 20% al 50% dei casi a esordio neurologico e può
essere di difficile trattamento.
1Scuola
di Specializzazione in Pediatria, Università di Modena e
Reggio Emilia
2Pediatria,
Ospedale Santa Maria Nuova, Reggio Emilia
3U.O.
Pediatria, Dipartimento Integrato Materno-Infantile, Az. Osp.
Universitaria, Policlinico di Modena
A., 11
anni, giunge presso il PS per febbre da 10 giorni, non rispondente
alla terapia antibiotica. Comparsa di esantema diffuso da alcuni
giorni. Obiettivamente faringe iperemico con zaffi di pus, esantema
al volto, tronco e arti, tumefazione della tibio-tarsica bilaterale e
della II-III-IV metacarpo-falangea sinistra. Gli esami evidenziano
leucocitosi neutrofila e rialzo degli indici di flogosi, all'rx
torace piccolo focolaio in sede retrocardiaca con versamento pleurico
e pericardico. Si imposta copertura antibiotica con ceftazidime e si
ricovera. In 2^ giornata riscontro di iperferritinemia (43.000 ng/ml)
con persistenza di esantema fisso e artralgie, nel sospetto di sdr da
attivazione macrofagica (MAS) in AIG sistemica si inizia terapia con
boli di metilprednisolone con transitoria riduzione delle tumefazioni
articolari e dell'esantema. Nei giorni successivi, presenza di
segni clinici e di laboratorio di ripresa di malattia per cui si
avviano, in successione, ciclosporina ev e inibitore TNF alfa, con
beneficio transitorio. Decidiamo quindi di iniziare terapia con
antagonista dell'IL-1, sospendendo l'anti TNF e iniziando
décalage della ciclosporina con progressivo miglioramento
clinico. La MAS è una complicanza potenzialmente fatale
dell'AIG sistemica causata da un' alterazione nei meccanismi di
presentazione dell'antigene, rilascio di citochine proinfiammatorie
e attivazione dei macrofagi. La MAS può presentarsi
all'esordio della malattia o come sua complicanza, il
riconoscimento precoce permetterebbe di ridurre la mortalità
che resta tutt'oggi elevata. Numerosi sintomi della MAS (febbre,
linfoadenopatia, epatomegalia, esantema) sono presenti anche nell'AIG
non complicata da cui la difficoltà diagnostica. Recentemente
sono stati proposti criteri (caduta di PLT, Hb e GB,
iperferritinemia, presenza di macrofagi emofagocitici nel midollo,
aumento degli enzimi epatici, febbre continua persistente,
ipofibrinogenemia e ipertrigliceridemia) che permetterebbero di
elaborare uno score su cui basare la diagnosi.
S.C.
di Nefrologia e Dialisi, AORN Santobono-Pausilipon, Napoli
Le
infezioni da parvovirus B19 solitamente hanno un decorso asintomatico
o si estrinsecano con quadri clinici benigni e autolimitantesi quali,
quinta malattia, crisi aplastica transitoria, rash, artralgia e
artrite. In taluni casi, però, l'infezione determina un
coinvolgimento sistemico tale da mimare l'esordio di LES (presenza
di 3-5 criteri dell'American College of Rheumatology) che si
dissolve con la risoluzione dell'evento acuto, in altri ancora
l'infezione stessa scatena un quadro clinico e laboratoristico
persistente di LES. Sono stati segnalati pochi casi di uveite
associata a infezione da parvovirus B19, mentre tale condizione è
spesso presente in soggetti affetti da LES.
Obiettivo.
Descriviamo la storia di un'infezione da parvovirus B19
manifestatasi come TINU (Nefrite tubulo-interstiziale e uveite)
syndrome e che ha mimato un LES.
Case
report. Maschio, età 6 anni, si presenta con febbre,
nausea, cefalea e iperemia congiuntivale e dopo pochi giorni comparsa
di artralgie invalidanti agli arti inferiori. Pratica terapia
antibiotica senza effetto. Gli esami ematochimici evidenziano indici
infiammatori molto elevati, iperazotemia, ipoalbuminemia,
ipergammaglobulinemia, piastrinosi e anemia. La visita oculistica
mostra uveite anteriore bilaterale per la quale inizia terapia
steroidea sistemica e locale. Viene posta la diagnosi di TINU
syndrome. Le indagini mostrano: presenza di IgG e IgM anti-parvovirus
B19; ANA e DNAasi positivi; ANCA, anti ds-DNA, AMA, ASMA e LAC
negativi; C3 e C4 ridotti; Test di Coombs diretto positivo (IgG +1).
L'esame delle urine evidenzia microematuria ma non proteinuria.
L'Ecografia renale segnala reni iperecogeni. La settimana
successiva la funzionalità renale si è
sorprendentemente normalizzata e la visita oculistica non ha più
mostrato l'uveite. Dopo qualche settimana si è assistito
anche alla negativizzazione degli indici laboratoristici di LES e
infine un mese dopo sono scomparse le IgM anti-parvovirus B19.
Discussione.
In letteratura sono stati descritti 15 casi di infezione da
parvovirus a esordio LES-like e 10 casi di infezione da parvovirus
che ha scatenato l'insorgenza di LES1,2. Dei 15 casi di
infezione a esordio LES-like solo uno si è presentato in età
pediatrica3. Queste evidenze supportano la teoria che
nella patogenesi del LES l'infezione da parvovirus possa fungere da
fattore triggering. Studi ulteriori saranno necessari per comprendere
quali siano i fattori che determinano la differente evoluzione
clinica (remissione del quadro clinico-laboratoristico LES-like o
persistenza dello stesso).
Conclusioni.
Il nostro è l'unico caso riportato di TINU syndrome
associata a infezione da parvovirus B19 simulante un LES. Il
messaggio che vogliamo lasciare è che anche se l'uveite può
far parte del pattern sintomatologico-clinico delle malattie
autoimmuni, riteniamo che prima di attribuirla a una causa puramente
autoimmune va sempre esclusa l'infezione da parvovirus B19 quale
eziologia di una malattia così complessa.
Bibliografia
- Aslanidis S, et al. Parvovirus B19 infection and systemic lupus erythematosus: Activation of an aberrant pathway? Eur J Intern Med 2008;19:314-8.
- Hession MT, et al. Parvovirus B19-associated systemic lupus erythematosus: clinical mimicry or autoimmune induction? J Rheumatol 2010;37:2430-2.
- Watanabe Y, et al. Self-limited lupus-like presentation of human parvovirus B19 infection in a 1-year-old girl. Pediatr Int 2009;51:411-2.
Pediatria
D'Urgenza, Ospedale Infantile Regina Margherita, Torino
Il
rachitismo, raro alle nostre latitudini, è in aumento nelle
minoranze etniche per il fenomeno immigratorio1. Le cause principali
sono date dalla mancata esposizione alla luce solare e
dall'insufficiente apporto di calcio e vitamina D, in particolare
in caso di allattamento materno esclusivo1,2.
L'ipocalcemia è frequentemente asintomatica, ma le forme
gravi possono esitare in tetania, laringospasmo o convulsioni
generalizzate. La bradicardia e l'arresto cardiaco sono rari, ma è
possibile riscontare un allungamento del QT all'ECG3.
Descriviamo
il caso di una bambina egiziana di 15 mesi, condotta in P.S. per
rigidità diffusa specie alle estremità distali degli
arti in corso di gastroenterite. Restante obiettività
negativa. Si riscontravano ipocalcemia severa, ipofosforemia,
ipomagnesemia, aumento della fosfatasi alcalina e incremento del
paratormone. ECG nella norma. Per l'impossibilità di
reperire un accesso venoso periferico si procedeva al posizionamento
di un accesso intraosseo per l'infusione di calcio gluconato (2
ml/kg/die) con regressione della sintomatologia in circa 3 ore. a una
più attenta ispezione si evidenziavano alcune delle tipiche
stigmate rachitiche. La bambina, alimentata esclusivamente con latte
materno sino a 8 mesi, non aveva mai effettuato supplementazione con
vitamina D. Questo, in associazione a una bassa esposizione ai raggi
solari per motivi etnico-culturali, è responsabile
dell'ipocalcemia, verosimilmente precipitata dall'episodio
gastroenteritico. Si sottolinea l'utilità dell'accesso
intraosseo per una pronta terapia in caso di impossibilità di
reperire un accesso venoso periferico in condizioni a rischio per la
vita.
Bibliografia
- Allgrove J. Is nutritional rickets returning? Arch Dis Child 2004;89:699-701.
- Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest 2006;116:2062-72.
- Behrman RE, et al. Nelson Textbook of Pediatrics, XVIII ed.
Clinica
Pediatrica, Servizio di Allergologia, IRCCS pediatrico Burlo
Garofolo, Trieste
Riportiamo
la casistica dei casi di anafilassi idiopatica afferiti presso il
Servizio di Allergologia dell'Ospedale Burlo Garofolo di Trieste
degli ultimi 3 anni.
L'anafilassi
idiopatica (AI), definita come anafilassi senza possibile
identificazione di causa precipitante, è un'entità
clinica rara, soprattutto in età pediatrica. Alla base del
disturbo è possibile via sia una patogenesi autoimmune;
clinicamente si manifesta con orticaria, angioedema, ipotensione,
tachicardia, asma, prurito, nausea, vomito, diarrea fino alla perdita
di coscienza e colpisce tipicamente l'età giovane-adulta. Va
da sé che l'AI è una diagnosi d'esclusione, laddove
non è possibile identificare una precisa causa scatenante
l'anafilassi; le diagnosi differenziali sono poche (mastocitosi,
sindrome da carcinoide, deficit di C1q inibitore, feocromocitoma).
Negli
ultimi 3 anni sono afferiti al nostro centro 9 pazienti con diagnosi
di AI. L'età media alla diagnosi è stata di 12 anni
(range 6-18 anni). Soltanto 1 caso ha manifestato angioedema isolato,
in tutti gli altri casi venivano riportati anche sintomi sistemici:
più frequentemente asma, diarrea, angioedema, in un solo caso
ipotensione con tachicardia, nessuno aveva presentato perdita di
coscienza. In 5 casi poteva definirsi una AI di tipo infrequente, i
restanti casi erano a presentazione frequente. Il tempo di sviluppo
dei sintomi era nella maggior parte dei casi rapido (15 min), in 3
casi la clinica completa poteva presentarsi fino a 6 ore dall'inizio.
In tutti i casi era stata prescritta l'adrenalina IM per
autoinoculazione (tranne nel caso a forma localizzata) e una terapia
profilattica con antistaminico.
In
conclusione la descrizione della nostra casistica è abbastanza
in linea con i dati riportati dalla letteratura corrente, malgrado
poche siano le casistiche pediatriche riportate: 1. l'età
d'insorgenza spostata verso la pubertà; 2. la
modalità clinica di presentazione: stereotipata nei diversi
episodi, che non mette mai in pericolo di vita e autolimitantesi; 3.
la terapia profilattica: riduce la frequenza degli episodi e ne
attenua la modalità di presentazione; 4. una malattia
che, malgrado la gravità di presentazione, va sempre a buon
fine.
Clinica
Pediatrica, Servizio di Gastroenterologia, IRCCS pediatrico Burlo
Garofolo, Trieste
La
videocapsula è la prima tecnica di imaging capace di
visualizzare in vivo la mucosa del piccolo intestino. Si conosce la
possibilità di utilizzare la videocapsula nella diagnostica
del diverticolo di Meckel ma in letteratura sono segnalati solo
report di casi singoli e casi appartenenti a case series. Abbiamo
analizzato i dati dei pazienti ricoverati presso nostra Clinica
Pediatrica con diagnosi di diverticolo di Meckel dal gennaio 2009 a
febbraio 2011.
La
casistica comprende 8 casi, 5 maschi e 3 femmine. In 3 casi l'esordio
è stato prima dei 2 anni, in 2 prima dei 4 anni, 1 a 7 anni e
2 casi dopo i 10 anni. L'emorragia intestinale è stato il
sintomo d'esordio più frequente, presente in 6 bambini, in 1
solo caso si è manifestato con dolore addominale non associato
ad altri sintomi e in 1 caso con anemia sideropenica isolata. In 7
hanno eseguito l'esame con videocapsula, 6 di questi hanno eseguito
anche la scintigrafia: in 2 casi entrambi gli esami sono risultati
positivi, in 1 caso la scintigrafia non è stata eseguita
perché la clinica di crisi subocclusive e il reperto della
videocaspula hanno portato subito all'intervento in laparoscopia e
in 1 caso per età e clinica (enteroraggia e anemizzazione
acuta) il bambino è stato sottoposto direttamente a
laparoscopia.In 5 su 7 casi la videocapsula ha visualizzato immagini
compatibili con diverticolo (tutti confermati alla laparoscopia), in
3 casi su 6 la scintigrafia è risultata positiva (tutti
confermati).
In un
solo caso entrambi gli esami sono risultati negativi.
Dei 2
casi negativi alla videocapsula uno era risultato positivo alla
scintigrafia e uno negativo; in quest'ultimo caso la diagnosi è
stata fatta in laparoscopia. In 2 dei 3 casi negativi alla
scintigrafia la videocapsula ha permesso la diagnosi.
Conclusione.
La diagnosi di diverticolo di Meckel era e rimane una diagnosi
difficile. La videocapsula si aggiunge alla scintigrafia dimostrando
una maggiore sensibilità e può essere considerato come
esame di prima scelta quando vengano eseguite contestualmente EGDS e
colonscopia in sedazione, senza aggiungere invasività e senza
effetti collaterali, anche in bambini molto piccoli. La specificità
dell'immagine è molto elevata, in tutti i casi il reperto
della videocapsula è stato confermato all'intervento
chirurgico.
Peraltro
la sensibilità dell'esame nella nostra piccola casistica non
è assoluta (88,9%); tale dato, assieme ai costi elevati (costo
per videocapsula: 600 euro) e al fatto che rimane un esame eseguibile
solo in Centri di endoscopia pediatrica di terzo livello, non
permette di considerare la videocapsula esame di prima scelta in
tutti i casi di sospetto diverticolo di Meckel.
Quindi,
seppur l'associazione dei 2 esami permetta la diagnosi nella
maggioranza dei casi (6 su 7 nella nostra casistica), in caso di
forte sospetto clinico, con videocapsula e scintigrafia negative,
rimane l'indicazione alla esplorazione laparoscopica.
8
casi |
Positivo |
Negativo | |
Videocapsula |
7 |
5 |
2 |
Scintigrafia |
6 |
3 |
3 |
Nessun esame |
1 |
/ |
/ |
Clinica
Pediatrica, IRCCS pediatrico Burlo Garofolo, Trieste
Introduzione.
Il dolore è un problema rilevante per i bambini con deficit
cognitivo, ma la sua misurazione è spesso difficoltosa.
L'obiettivo del nostro studio è quello di confrontare le
qualità di 3 diverse scale osservazionali per la valutazione
del dolore, di cui due specifiche per bambini con ritardo mentale
(NCCPC-PV e DESS) e una non specifica (CHEOPS).
Materiali
e metodi. 20 bambini con deficit cognitivo, non comunicanti, sono
stati valutati, nel corso di uno stimolo doloroso, impiegando le 3
scale sopra indicate. Ogni valutazione è stata effettuata, da
due osservatori esterni e da un genitore, contemporaneamente e in
cieco. Per ogni scala sono state analizzate riproducibilità
interoperatore, concordanza con i risultati delle altre scale,
semplicità d'uso, oggettività, e accuratezza.
Risultati.
le due scale specifiche (NCCPC-PV e DESS) hanno dimostrato una
migliore coerenza nella discriminazione del dolore rispetto alla
CHEOPS. La DESS ha mostrato la migliore riproducibilità
interoperatore. La NCCPC-PV è risultata la scala più
oggettiva perché più indipendente dalla conoscenza del
bambino, inoltre è stata ritenuta la più semplice da
usare dagli osservatori esterni e secondo i genitori è la
scala che meglio descrive il dolore nel proprio bambino.
Discussione.
Per misurare correttamente il dolore nei bambini con deficit
cognitivo severo è necessario utilizzare degli strumenti
algometrici osservazionali specifici e validati. La NCCPC-PV è
uno strumento adeguato per l'utilizzo nella pratica clinica, perché
soddisfa le necessarie caratteristiche di validità,
riproducibilità, semplicità d'uso, accuratezza e
oggettività.
U.O.
di Pediatria, Ospedale Bufalini, Cesena;
Scuola di specializzazione in pediatria, IRCCS pediatrico Burlo Garofolo, Università di Trieste
Scuola di specializzazione in pediatria, IRCCS pediatrico Burlo Garofolo, Università di Trieste
Premessa.
La gestione del bambino con diabete a scuola è stata finora
esclusivamente a carico delle famiglie. La somministrazione della
terapia insulinica da parte del personale scolastico è spesso
rifiutata per il timore di somministrare una dose errata di insulina
oltre che per la reticenza nel praticare l'iniezione stessa. Ciò
comporta spesso la rinuncia da parte delle famiglie a far mangiare il
bambino alla mensa scolastica e, spesso, alla partecipazione alle
gite scolastiche.
Un
questionario precedentemente somministrato a 30 famiglie con bambini
con diabete dal nido alle scuole elementari afferenti al centro
diabetologico di Cesena aveva evidenziato il bisogno da parte delle
famiglie che sia il personale scolastico a somministrare l'insulina
durante le ore scolastiche.
Peraltro
un recente decreto legge promulgato nella regione Emilia Romagna
sancisce il diritto da parte del bambino con patologia cronica di
ricevere la somministrazione dei farmaci necessari in orario
scolastico da parte del personale scolastico.
Obiettivo.
riportare l'esperienza del centro diabetologico di Cesena nella
gestione a scuola del bambino con diabete.
Materiali
e metodi. Abbiamo raccolto l'esperienza di 15 bambini con
diabete che frequentano il nido, la scuola materna e la scuola
elementare, di cui 10 con microinfusore e 5 in terapia
multi-iniettiva. Tutti i bambini mangiano alla mensa scolastica.
1.
Gestione del bambino con microinfusore
I bambini
ricevono la stessa dieta dei coetanei. La dietista fornisce il
calcolo dei carboidrati frazionato per alimento relativo al vassoio
offerto. Il personale scolastico con l'ausilio del calcolo fornito
dalla dietista e del microinfusore che fornisce il calcolo automatico
del bolo insulinico da erogare, somministra l'insulina al pasto nel
bambino più piccolo o supervisiona la gestione del
microinfusore nel bambino più grande. In questo modo il
personale scolastico è facilitato nel decidere la dose di
insulina da somministrare e non deve praticare direttamente
l'iniezione. L'utilizzo del microinfusore permette inoltre
l'eventuale frazionamento dell'insulina somministrata,
particolarmente utile nel bambino piccolo in cui non è sempre
possibile stabilire a priori la quantità esatta di cibo che
mangerà al pasto.
2.
Gestione del bambino senza microinfusore
Il
posizionamento dell'ago del microinfusore per la somministrazione
dell'insulina permette di evitare le iniezioni dirette da parte del
personale scolastico, facilitando la somministrazione dell'insulina
al bambino.
Conclusioni.
L'impiego delle nuove tecnologie aiuta le famiglie nella gestione
del bambino con diabete a scuola, permette al bambino una vita
sociale uguale a quella dei coetanei senza limitazioni legate alla
sua patologia e aiuta gli insegnanti a superare i timori legati alla
gestione di questa problematica.

SESSIONE
POSTER
Servizio
di Oncoematologia Pediatrica e patologie della coagulazione, Ospedale
Regionale per le Microcitemie, Cagliari
Il
passato quasi remoto. 5 anni fa escissione di neoformazione a
carico del palmo della mano sinistra, diagnosi: sarcoma a cellule
chiare (non ripetizioni metastatiche, massa localizzata e chirurgia
radicale). Vista la scarsa chemio-radiosensibilità del tumore
e la localizzazione singola di malattia, si decide per follow-up
clinico strumentale. Il ragazzo presenta ottime condizioni generali,
pratica attività sportiva agonistica (ballo tip tap) e le
rivalutazioni di malattia sono sempre risultate negative.
Il
passato ritorna? A 5 anni dalla chirurgia, A. presenta forti
dolori a carico del bacino scarsamente responsivi alla terapia
antidolorifica e anti infiammatoria; la Rx del bacino è
negativa per lesioni ossee.
Per la
persistenza della sintomatologia dolorosa viene sottoposto a
ecografia dell'addome che mostra la presenza di voluminosa massa a
livello della pelvi e fossa iliaca sinistra. Esegue quindi TC che
evidenzia a sinistra in sede retroperitoneale pelvica estesa sino
allo spazio sottoperitoneale grossolana formazione solida a margini
netti, disomogenea di 151 x 110 mm che comprime e disloca verso
destra il muscolo psoas. Il muscolo gluteo medio e il muscolo iliaco
sinistro appaiono compressi e infiltrati.
Il
caso. A. si presenta alla nostra attenzione dopo l'esito degli
esami strumentali. Il problema, una volta confermati gli esiti degli
esami strumentali ed eseguita una valutazione clinica completa delle
condizioni generali e del performance status del paziente è la
definizione della natura della neoformazione. Si tratta di una
recidiva a distanza o di una seconda neoplasia maligna?
Le
valutazioni multidisciplinari (consulenza chirurgica, BM, biopsia con
true-cut, centralizzazione dei campioni istologici con analisi delle
traslocazioni e valutazione in biologia molecolare) hanno permesso di
dirimere il quesito clinico.
La
risposta. Neoplasia tipo PNET/EWING con traslocazione t(11,22)
evidenziata a carico della massa e dell'aspirato midollare cresta
iliaca sinistra. Il paziente è stato arruolato al protocollo
EpSSG RMS 2005. Quindi il paziente ha un altro tumore.
La
nuova domanda. Il midollo risulta essere positivo per
localizzazione di malattia? Si tratta di una positività
secondaria all'infiltrazione da parte della massa?
I
problemi. Valutazione precisa dell'istologia tumorale;
definizione puntuale dello stadio di malattia in relazione alla
possibile invasione midollare.
La
soluzione. È stato possibile individuare la corretta e
precisa istologia tumorale grazie alla stretta collaborazione con
l'anatomopatologo e alla centralizzazione dei campioni come da
mission AIEOP.
Il
contributo. Essenziale è stato il contributo dato dagli studi
di biologia molecolare. Siamo tutt'ora in attesa dei risultati
delle valutazioni comparative che ci permetteranno di esplorare in
maniera più completa la possibile presenza di malattia anche a
livello midollare.
UO
Oncoematologia pediatrica, IRCCS pediatrico Burlo Garofolo,
Trieste
L. è
un bambino di 11 anni con linfadenopatia diffusa da circa 2 mesi,
inizialmente laterocervicale e retro auricolare bilaterale, poi
estesa anche alla regione sovraclaveare sinistra, con incostante
dolenzia locale. Non ci sono stati sintomi/segni sistemici (febbre,
calo ponderale, sudorazione notturna, astenia). All'ecografia si
visualizzavano pacchetti linfonodali in cui alcuni presentavano
vascolarizzazione ilare, senza sovvertimento della struttura
linfonodale, altri senza segni di vascolarizzazione alcuna, di dubbio
significato. Negativi erano risultati emocromo, VES, PCR, LDH,
negative sierologia per bartonella, EBV, CMV, toxoplasma, rosolia,
tularemia, Mantoux, test al superossido e radiografia del torace nel
sospetto di linfoma diffuso; nella norma le sottopopolazioni
linfocitarie su sangue periferico a più riprese, nel sospetto
di leucemia. In attesa di eseguire una biopsia linfonodale per
approfondire il quadro, si notavano segni di infiammazione con
spiccata dolenzia di alcuni linfonodi, per cui nel sospetto di
linfadenopatia infettiva veniva avviata terapia antibiotica
(inizialmente amoxicillina/clavulanato, poi azitromicina e
ciprofloxacina, pensando a una forma di micobatteriosi atipica) con
risposta solo parziale: la successiva biopsia linfonodale aveva dato
risultati non dirimenti, per la presenza di necrosi estesa senza
cellule tipizzabili; negativa la coltura per micobatteri atipici.
Considerando il peggioramento della linfoadenopatia con indagini
persistentemente negative, se non per modesto rialzo degli indici di
flogosi, è stata eseguita una seconda biopsia linfonodale a
distanza di 3 settimane: in questa occasione è stata rilevata
l'infiltrazione linfonodale da parte di blasti pre-B, presenti
anche su sangue periferico (che era negativo in tal senso una
settimana prima), con emocromo ancora normale. L'aspirato mostrava
invasione (80%) di linfoblasti L1. È stata così fatta
diagnosi di leucemia linfoblastica acuta, pur con emocromo sempre
normale, con notevole linfoadenopatia come primo segno clinico, forse
anche da linfadenite infettiva necrotizzante facilitata dalla
immunodepressione.
Scuola
di specializzazione in Pediatria, Università di Trieste
E. è
una bambina di 2 anni e dieci mesi, seguita presso il nostro Istituto
perché affetta da Neurofibromatosi di tipo 1, diagnosticata
all'età di sei mesi per la presenza di macchie caffèlatte,
efelidi ascellari e inguinali. Viene alla nostra attenzione per la
comparsa da qualche giorno di strabismo convergente dell'occhio
sinistro, atteggiamento obbligato del capo verso destra e andatura
barcollante. Nei pazienti affetti da neurofibromatosi di tipo 1, in
cui si manifestino sintomi neurologici e oculari deve sorgere il
sospetto di glioma del nervo ottico e si deve pertanto procedere con
l'esecuzione di una risonanza magnetica encefalica. Nel caso di E.
l'indagine ha confermato la presenza di una lesione rotondeggiante
captante contrasto lungo il decorso delle vie ottiche di destra,
reperto compatibile con il glioma delle vie ottiche. Il glioma del
nervo ottico è presente in circa il 15% dei pz affetti da NF1
e la maggior parte sono asintomatici, tuttavia nel 20% dei casi
possono verificarsi delle complicanze. Lesioni di questo tipo
richiedono un monitoraggio di tipo clinico-strumentale (comparsa di
segni di ipertensione endocranica, alterazioni del visus e della
motilità oculare, proptosi, deficit di campo visivo
periferico, pubertà precoce) che consenta un trattamento
tempestivo in caso di accrescimento della lesione. La presenza di
sintomi costituisce indicazione al trattamento. È
sconsigliabile il ricorso alla radioterapia, dati gli effetti
teratogeni, ancor più temibili in un paziente con alterata
funzionalità onco-soppressoria. Anche l'approccio
chirurgico, di per sé difficoltoso per lesioni del sistema
nervoso centrale, risulta prognosticamente sfavorevole data l'elevata
incidenza di recidiva a causa della reazione cellulare infiammatoria
post-intervento. Per E. quindi, è stata avviata la
chemioterapia con vincristina e carboplatino.
Conclusioni.
In presenza di segni oculari suggestivi di glioma ottico (es.
strabismo) in pazienti affetti da NF1, è indicato eseguire una
risonanza magnetica dell'encefalo. La diagnosi tempestiva di tale
lesione consente di avviare immediatamente il trattamento
chemioterapico, migliorandone la prognosi.
Clinica
Pediatrica, IRCSS pediatrico Burlo Garofolo, Trieste
G. è
un ragazzo di 14 anni che viene in PS per la comparsa improvvisa di
cefalea frontale insorta durante un allenamento di tennis che lui
pratica a livello agonistico. La cefalea non era migliorata con
l'assunzione di paracetamolo ma era scomparsa autonomamente. Un
secondo episodio era iniziato dopo aver fatto le scale a scuola.
Numerose visite sportive non avevano mai evidenziato problemi, ma la
pressione arteriosa misurata sia dal curante sia a domicilio era
risultata elevata (sistolica 150 mmHg, diastolica 90 mmHg).
All'ingresso in PS il ragazzo si presentava in ottime condizioni
generali, con obiettività cardiaca e neurologica negative e
normali valori pressori. Lo stix urine documentava una modesta
proteinuria senza altre anomalie. La cefalea scatenata dalla sforzo,
l'episodica ipertensione e la proteinuria ci hanno indotto ad
approfondire gli accertamenti diagnostici. Fundus oculi e
funzionalità renale sono risultati nella norma, mentre
all'ecografia addominale è stata visualizzata una lesione
ipoecogena extravasale di circa 2 cm di diametro che comprimeva
parzialmente i vasi renali. Per sede e morfologia è stato
escluso si potesse trattare di una tumefazione linfonodale. È
stata quindi eseguita una TC dell'addome per valutare meglio la
natura della massa.
La TC
addominale ha confermato il reperto ecografico mostrando una
formazione ovalare, solida, localizzata posteriormente alla
confluenza della vena renale destra nella cava inferiore. ECG,
radiografia del torace e RMN encefalo erano nella norma. Nel sospetto
di feocromocitoma è stato fatto il dosaggio delle catecolamine
urinarie, che ha evidenziato valori di adrenalina urinaria 20 volte
maggiori di quelli normali. Per escludere ulteriori localizzazioni è
stata eseguita una scintigrafia con MIBG risultata negativa. La
diagnosi definitiva ci è stata fornita dall'esame istologico
della massa asportata chirurgicamente: si trattava di un
paraganglioma, neoplasia che, come il feocromocitoma, origina dalle
cellule cromaffini di origine neuroectodermica; la sua localizzazione
è extra-surrenalica. Queste neoplasie secernono catecolamine:
la loro tipica manifestazione clinica è quindi l'ipertensione,
che può essere stabile o a crisi parossistiche; lo sforzo
fisico è un classico trigger.
Oncoematologia,
IRCSS pediatrico Burlo Garofolo, Trieste
C. è
una bambina di 7 anni che viene inviata al nostro Centro di
Oncoematologia per una piastrinopenia, riscontrata 3 anni prima nel
corso di accertamenti pre-operatori per intervento chirurgico
correttivo per displasia dell'anca congenita. La piastrinopenia non
aveva risposto a cortisonici e IVIG, l'esame del midollo osseo
aveva evidenziato un midollo ipoplastico, senza note di
mielodisplasia o presenza di cellule anomale. La piastrinopenia si
era confermata in successivi controlli e aveva impedito un nuovo
tentativo di intervento a distanza di alcuni anni dal primo.
Gli esami
eseguiti alla valutazione degli ematologi mostrano una significativa
piastrinopenia (36.000 piastrine/mm3), lieve anemia (Hb 11,6 g/dl)
macrocitica (MCV 102 fl) e neutropenia (1300 N/mm3), un aumento
dell'HbF (4,7%). C., oltre all'importante displasia dell'anca
destra, ha una facies particolare: taglio orientaleggiante degli
occhi, lieve ipertelorismo e micrognazia. È inoltre piccola di
statura, anche se nella sua classe, ricca di bambini stranieri, non è
la più bassa. La sua crescita staturo-ponderale è al di
sotto delle 3 DS e due test del GH con stimolazione risulteranno
patologici. Infine all'ecografia addome viene riscontrata la
presenza di una loggia renale destra disabitata con rene destro
pelvico.
La
biopsia osteomidollare ha documentato un quadro di sindrome
mielodisplastica; DEB test e test di complementazione hanno
confermato il sospetto diagnostico di anemia di Fanconi. È
stata avviata l'analisi genetica e la ricerca di un donatore di
cellule staminali ematopoietiche.
L'anemia
di Fanconi è una malattia rara, caratterizzata da fragilità
cromosomica. L'aplasia midollare è progressiva, con esordio
all'età media di 8 anni: di norma l'anemia è
l'ultimo segno ematologico a comparire, preceduto da piastrinopenia
e neutropenia. Sono però suggestivi l'elevato MCV e
l'aumento dell'HbF. Al quadro midollare si associano (in meno
della metà dei casi) dimorfismi, anomalie anatomiche, disturbi
endocrinologici, ritardo mentale (nel 10% dei casi). Vi è un
aumento dell'incidenza di neoplasie ematologiche e solide. L'unica
terapia è il trapianto di midollo allogenico, che risolve però
solo il problema ematologico, non modificando l'incidenza di
neoplasie e gli altri aspetti sindromici qualora presenti.
1Scuola
di Specializzazione in Pediatria, Università di Modena e
Reggio Emilia
2U.O.
Pediatria, Ospedale Santa Maria della Misericordia, Reggio Emilia
3S.C.
Chirurgia Pediatrica, Az. Ospedaliero-Universitaria, Parma
P., 12
aa, giungeva alla nostra osservazione per sintomatologia
caratterizzata da nausea e vomito da otto giorni, associata a puntate
febbrili (TC max 39,6 °C), alcuni episodi di alvo scomposto con
dolore addominale precedente l'evacuazione. Al quinto giorno di
febbre veniva visitata dal curante che, per il riscontro di flogosi
delle alte vie aeree, avviava terapia antibiotica con cefixime 400
mg/die. In decima giornata per il persistere di dolore addominale
crampiforme e sporadici episodi di vomito la ragazza veniva condotta
in PSP dove si riscontrava addome diffusamente dolente alla
palpazione addominale e profonda, con iniziale resistenza ai
quadranti inferiori. Si eseguivano esami ematici urgenti con rilievo
di spiccata leucocitosi neutrofila e aumento della PCR (PCT
negativa). L'ecografia addome mostrava in sede centrale espanso
sacciforme del diametro di circa 6 cm, a contenuto corpuscolato
livellato e la TC addome urgente Douglas impegnato da
formazione capsulata a densità idrica con micro bolle aeree di
7 x 5 cm; analoga formazione dotata di ampio livello posta
all'imbocco dello scavo pelvico di 8 x 7 cm. Terza formazione
solida che si aggetta tra le due nel cui contesto si riconosce quello
che potrebbe essere riferibile a gemma dentaria. Il quadro può
essere compatibile con cisti dermoide, la presenza di gas con
complicanza flogistica. Nel sospetto di teratoma multiplo
complicato da sovrainfezione batterica si assisteva a un netto
incremento del dolore addominale, con crisi algiche a carattere
colico, associato a grave stato di sofferenza generale con aspetto
settico della paziente, pertanto la ragazza veniva trasferita
d'urgenza in chirurgia pediatrica. Soltanto sul tavolo operatorio
si poteva constatare che le tre neoformazioni facevano parte di un
unico ascesso saccato a carico dello scavo pelvico che si estendeva
fino all'ombelicale trasversa, con cotenna fibrosa coinvolgente
utero e annessi, configurando così un quadro di appendicite
saccata con sintomatologia mascherata dalla terapia antibiotica
intrapresa. Nonostante ciò, in sede d'intervento, si
riusciva a salvaguardare le strutture pelviche coinvolte. Il decorso
post-operatorio risultava regolare.
U.O.
Neonatologia, Dipartimento Materno-Infantile, AOUP Pisa
1U.O.
Pediatria 2, Dipartimento Materno-Infantile, AOUP Pisa
Caso
clinico. G. nasce a 34 settimane di EG con TC per IUGR. Primi
atti fisiologici nella norma, peso < 10° centile (1755 g). La
neonata presenta diverse manifestazioni cutanee ipercromiche
lievemente infiltrate e variamente distribuite. Eco cerebrale, EEG,
eco addominale e visita oculistica nella norma. La consulenza
dermatologica conferma la presenza di 2 lesioni papulo-nodulari su
superficie anteriore gamba sx, 2 lesioni simmetriche in regione
lombare e 1 lesione maculare su palmo mano dx. Nel sospetto di
istiocitosi X vengono eseguiti biopsia ed esame istologico di una
delle lesioni che dimostrano reperti compatibili con localizzazione
cutanea di istiocitosi a cellule di Langerhans (istiocitosi X)
(grandi istiociti mononucleati positivi per S100, CD1a, LCA). La
progressiva regressione spontanea delle lesioni ha permesso uno
stretto follow-up esclusivamente clinico che continua a tutt'oggi.
Discussione.
I disordini istiocitici, tra cui l'istiocitosi X è la
forma più comune, sono un gruppo eterogeneo di patologie
caratterizzato da un'anomala proliferazione monoclonale e da un
accumulo di istiociti reattivi in vari organi e tessuti. Le
caratteristiche della malattia suggeriscono un processo disregolativo
di tipo immune, reattivo o neoplastico. Le manifestazioni più
comuni sono le lesioni ossee (cranio e femore prossimale); possibile
l'interessamento di cute e mucose (55%), SNC (35%), sistema
epatobiliare e milza (32%), polmone (26%) e altro. L'incidenza è
1/1.000.000 di bambini con età < 15 aa, con prevalenza nei
maschi. Nel bambino con età < 1 anno l'organo più
colpito è la cute nelle sedi inguinale, lombosacrale,
ascellare, del capo e del collo e le lesioni possono essere nodulari,
maculo-papule eritematose e crostose o xantomi papulari. La diagnosi
si basa su riscontro bioptico di tipiche cellule di Langerhans con
positività immuno-istochimica per S100, CD1a e LCA. Spesso la
prognosi è buona con regressione spontanea delle lesioni;
nelle forme a evoluzione progressiva con interessamento sistemico si
può ricorrere a chemioterapia o immunosoppressione. Take home
message. Nei bambini le lesioni cutanee possono rappresentare l'unica
manifestazione dell'istiocitosi X.
SS
Gastroenterologia, Epatologia e Nutrizione Clinica, IRCCS pediatrico
Burlo Garofolo, Trieste
1Scuola
di Specializzazione in Pediatria, Università di Trieste
Background.
L'ileoscopia mediante videocapsula (VC) permette lo studio non
invasivo dell'intestino tenue ed è una tecnica facilmente
accettata dagli adulti e dai bambini in grado di ingoiare le capsula.
Pochi sono i dati relativi ai bambini più piccoli nei quali la
VC deve essere introdotta in corso di esofagogastroduodenoscopia
(EGDS).
Obiettivi.
Valutare le indicazioni e la sensibilità diagnostica della VC
nei bambini di età inferiore ai 10 anni.
Metodi.
Sono stati raccolti i dati relativi ai pazienti di età <10
anni sottoposti a posizionamento della capsula mediante applicatore
Advance in corso di EGDS; sono state raccolte le indicazioni
all'esecuzione dell'esame, le lesioni rilevate e la diagnosi di
dimissione.
Risultati.
Abbiamo individuato 37 pazienti (23 M, 14 F) per un totale di 39 VC
posizionate. In 38 casi la VC è stata posizionata oltre il
piloro; in 1 solo caso la capsula è stata posizionata nello
stomaco e non c'è stato passaggio nel duodeno (esame poi
ripetuto).In nessun caso vi sono state complicanze dovute alla
tecnica di posizionamento né ritenzione della VC, nonostante
lo studio dell'intestino tenue non sia stato preventivamente
eseguito in nessun caso. L'età media dei pazienti è
stata 5,96 anni (range 1,58-9,92), il peso medio 21,97 kg (range 7,50
-51,6). I pazienti distribuiti per classi d'età: < 2
anni: 1; 2-4 anni: 11; 4-6 anni: 7; 6-8 anni: 9; 8-10 anni: 11. Le
indicazioni all'esecuzione della VC sono state: sanguinamento
intestinale o anemia senza altra causa in 15 casi, sospetta o
confermata malattia infiammatoria cronica intestinale (MICI) in 16
casi (tutti avevano eseguito anche una colonscopia), dolori
addominali in 3 casi, poliposi intestinale in 3 casi, sospetta
linfangectasia intestinale e sospetta invaginazione in 1 caso
rispettivamente. La VC ha dato esito positivo 20/39 (51,28%); i falsi
negativi sono stati 3/39 casi (7,69%). In 12/39 casi (30,77%) la
negatività della VC ha permesso la diagnosi differenziale: ha
escluso 7 possibili morbi di Crohn e 2 possibili poliposi intestinali
mentre ha definito la diagnosi di rettocolite ulcerosa.
Conclusioni.
L'esame con VC è risultato sicuro nei bambini al di sotto
dei 10 anni. Il sanguinamento intestinale o l'anemia sono state le
indicazioni più frequenti, seguite dal sospetto o dal
controllo di MICI. La VC non si è dimostrata affidabile nella
diagnosi di diverticolo di Meckel risultando negativa in 3/7 esami
(42,85%) né nello studio dei dolori addominali, mentre ha
permesso di confermare o escludere la presenza di un morbo di Crohn
nei casi sospetti o dubbi.
Priorità
di salute negli adolescenti con malattia infiammatoria cronica
intestinale: pazienti e medici a confronto
Clinica
Pediatrica, IRCCS pediatrico Burlo Garofolo, Trieste
Obiettivi.
Valutare la concordanza tra le priorità assistenziali delle
malattie infiammatorie croniche intestinali (MICI) nel vissuto dei
pazienti adolescenti, dei pediatri gastroenterologi e degli
specializzandi in pediatria.
Materiali
e metodi. Un questionario redatto ad hoc recante 23 affermazioni
relative alle problematiche degli adolescenti con MICI è stato
somministrato a pazienti adolescenti, pediatri gastroenterologi e
specializzandi in pediatria. Nel corso di una visita di controllo
presso la S.C.O. di Gastroenterologia ai pazienti è stato
chiesto rispondere al questionario attribuendo un punteggio (da 1 a
5) a ciascuna affermazione a seconda dell'importanza di
quest'ultima nel proprio vissuto quotidiano. Pediatri e
specializzandi hanno risposto al questionario attribuendo un
punteggio a ogni affermazione a seconda del peso che ritenevano
questa avesse nella vita dei propri pazienti affetti da MICI.
Risultati.
Confrontando le risposte dei tre gruppi è emersa una
discordanza significativa tra la classifica dei pazienti e quella dei
pediatri (soltanto un elemento in comune tra le prime cinque
priorità) e in misura minore tra quella dei pazienti e degli
specializzandi (due elementi in comune tra le cinque priorità
assistenziali). La discordanza si evidenzia anche nel fatto che due
tra le cinque priorità dei pediatri non compaiono neppure
nelle prime dieci posizioni della classifica dei pazienti. Inoltre i
punteggi dei pediatri e quelli degli specializzandi sono risultati
mediamente più elevati rispetto a quelli attribuiti dai
pazienti.
Conclusioni.
Dal nostro studio emerge una significativa discordanza tra le
priorità assistenziali dei pazienti affetti da MICI e dei
pediatri gastroenterologi.
1Clinica
Pediatrica
2Servizio
di Gastroenterologia Pediatrica
IRCCS
pediatrico Burlo Garofolo, Università di Trieste
Introduzione.
L'esofagite eosinofila (EE) e la più comune causa di
disfagia in età pediatrica. Si manifesta con disfagia, food
impaction, pirosi restrosternale (senza risposta all'antiacido) e
frequente è l'associazione con l'atopia. Aspetti
endoscopici tipici sono la trachealizzazione dell'esofago, le
placche biancastre e un denso infiltrato eosinofilo all'istologia.
L'attuale approccio terapeutico prevede la dieta di eliminazione,
gli steroidi locali e sistemici e gli antileucotrienici
(montelukast).
Obiettivi.
Analizzare le caratteristiche di una popolazione di pazienti con EE
in terapia di mantenimento con il montelukast, valutandone la
risposta clinico-endoscopica.
Materiali
e metodi. Studio retrospettivo basato sull'analisi delle
cartelle cliniche e dei referti endoscopici delle EGDS eseguite alla
diagnosi e durante il follow-up di una popolazione di 19 bambini con
EE afferenti al Servizio di Gastroenterologia Pediatrica di Trieste
dal 2003 al 2010. Sono stati valutati i dati demografici, la
presentazione clinica, la terapia somministrata e la risposta
clinico-endoscopica. La risposta clinica è stata classificata
come completa (risoluzione completa dei sintomi senza recidive) o
parziale (miglioramento dei sintomi ma ricaduta durante la terapia).
La risposta istologica è stata classificata come totale
(infiltrazione eosinofila < 15 eosinofili per campo ad alto
potere) o parziale (riduzione del numero di eosinofili, con conta
totale > di 15 eosinofili).
Risultati.
Dei 19 pazienti 4 sono stati posti in dieta di esclusione e pertanto
esclusi dallo studio. I rimanenti 15 (rapporto M:F 12:3, età
media 10 anni, range 2-15) sono stati trattati con montelukast (5-10
mg/die) dopo una terapia iniziale con steroidi orali (prednisone 2
mg/kg/die in 2 pazienti) o topici (fluticasone fatto male
125-750 µg/die in 9 pazienti) o entrambi (4 pazienti). 5
pazienti hanno sospeso il montelukast dopo 17,4 mesi (range 3-24) per
la completa risposta clinica-endoscopica. I rimanenti 10 sono
tutt'ora in terapia dopo un periodo di follow-up medio di 22,6 mesi
(range 2-48). 1 paziente non ha avuto risposta clinico-endoscopica, 3
hanno avuto una risposta clinica ed endoscopica parziale e 6 una
riposta clinica completa (rispettivamente 3 con risposta istologica
completa e 3 parziale). Non sono stati segnalati effetti avversi
legati all'utilizzo del montelukast (follow-up medio 30,2 mesi).
Conclusioni.
Nella nostra esperienza il montelukast ha rappresentato un
trattamento valido e sicuro come terapia di mantenimento della EE.
Nuovi studi sono necessari per chiarirne l'efficacia e il rapporto
costo-beneficio rispetto alla terapia steroidea.
Utilità
del dosaggio delle IgG4 in corso di induzione specifica di tolleranza
nell'allergia alimentare severa
Clinica
Pediatrica, IRCCS pediatrico Burlo Garofolo, Università
di Trieste
Introduzione.
L'anafilassi è un problema non infrequente nella pratica
clinica del pediatra. Il gold standard nella prevenzione
dell'anafilassi è la dieta di esclusione dall'alimento
offendente, mentre l'approccio in emergenza, a reazione in atto,
prevede l'utilizzo dell'adrenalina. Una valida alternativa alla
dieta di esclusione sembra essere l' Induzione della Tolleranza
Orale Specifica o SOTI.
Materiali
e metodi. Il nostro è uno studio monocentrico
retrospettivo. La popolazione si compone di 88 bambini giunti presso
la clinica pediatrica dell'ospedale Burlo Garofolo da gennaio 2008
a marzo 2011 per eseguire la SOTI per il latte e per i quali sia
stato possibile reperire almeno 2 dosaggi di IgG4 e IgE per
alfalattoalbumina, betalattoglobulina e caseina. I dosaggi sono stati
effettuati al ricovero e alla dimissione (T1 e T2) e nel follow-up, a
6, 12 e 18 mesi (T3, T4, T5). L'outcome viene considerato positivo
quando la quota di latte tollerata è > 5 ml, negativo se il
latte tollerato è < 5ml.
Risultati.
Abbiamo osservato un incremento significativo dei livelli sierici
delle IgG4 nel periodo del follow-up e un concomitante decremento
delle IgE nei bambini sottoposti a SOTI. Inoltre esiste una
differenza statisticamente significativa tra i livelli di IgG4
all'inizio della SOTI e al follow-up, nei soggetti con outcome
positivo.
Discussione.
In corso di SOTI per latte, le IgG4 specifiche sono dosabili in
concentrazioni progressivamente crescenti, con differenza
statisticamente significativa tra la concentrazione all'avvio della
SOTI e i controlli a distanza. Il mancato incremento delle IgG4 nel
periodo del follow-up, potrebbe avere un valore predittivo del
fallimento della procedura, permettendo di indirizzare il bambino
verso altre forme di desensibilizzazione (ad es. Sublinguale).
Pediatra
AULSS 16 Regione Veneto
Viene
presentata la casistica personale di 11 pazienti pediatrici (9
femmine e 2 maschi) di età 2-13 anni che hanno manifestato
reazione simil-malattia da siero (serum sickness-like reaction, SSLR)
nel periodo 2006-2010. In nove si è trattato di reazione ad
antibiotico orale (cefaclor: 4 casi; amoxicillina + clavulanato: 4
casi; cefixima: un caso); un decimo caso è descritto a parte;
in una paziente con PTI la reazione è stata verso il rituximab
e.v. Sette pazienti hanno presentato un quadro classico di SSLR; 4
bambine una forma accelerata. In una l'esordio dei sintomi è
avvenuto dopo ben 24 giorni (forma tardiva). In due bambine la SSLR
si è ripetuta con lo stesso farmaco: una di loro (caso 8) è
passata dalla forma classica a quella accelerata, nell'altra (caso
10), il tempo di latenza è rimasto invariato. Si segnalano
altri due casi particolari: una bambina (caso 3) ha presentato per
due volte un rash da amoxicillina, prima di manifestare una SSLR
accelerata verso cefaclor; un'altra bambina (caso 2) ha manifestato
una singolare serie di reazioni decelerate, cioè con
intervallo libero via via più lungo tra assunzione di
antibiotico e reazione; dopo il terzo episodio di reazione, un TPO
con amoxicillina ha dato esito negativo pur dimostrando un
progressivo calo di C3.
Gen |
Età
(anni) |
Farmaco |
Latenza
(giorni) |
Quadro
clinico |
Note | |
1 |
F |
2 |
cefaclor |
10 |
Rash,
artrite |
Classica |
2 |
F |
2 |
amoxi-clavul. |
1^
dose |
Rash
cutaneo |
|
cefaclor |
4 |
Rash
cutaneo |
||||
amoxicillina |
8 |
Orticaria |
||||
3 |
F |
3 |
amoxi-clavul. |
8 |
Rash
cutaneo |
|
amoxicillina |
5-6 |
Rash
cutaneo |
||||
cefaclor |
4 |
Febbricola,
orticaria, artrite |
Accelerata | |||
4 |
F |
4 |
amoxicillina |
24 |
Rash,
artrite |
Tardiva |
5 |
M |
5 |
amoxi-clavul. |
4 |
Rash,
artrite |
Accelerata |
6 |
F |
6 |
cefaclor |
1 |
Rash,
artrite |
Accelerata |
7 |
M |
7 |
cefaclor |
7 |
Orticaria,
artrite |
Classica |
8 |
F |
9 |
rituximab |
14 |
Artralgia |
Classica |
rituximab |
6 |
Febbre,
orticaria-angioedema, artralgia |
Accelerata | |||
9 |
F |
9 |
amoxicillina |
9
|
Febbre,
orticaria, artrite |
Classica |
10 |
F |
13 |
amoxi-clavul. |
7
|
Orticaria,
artrite |
Classica |
amoxi-clavul. |
7
|
Orticaria,
artrite |
Classica | |||
11 |
F |
13 |
cefixime |
14
|
Proteinuria;
febbre, orticaria, artralgia |
Classica |
La
malattia da siero (serum sickness) è stata descritta per la
prima volta nel 1905 da Von Pirquet e Schick come reazione a siero di
cavallo iniettato in bambini contro la difterite. Le SSLR sono
reazioni a farmaci con lo stesso meccanismo: mediate da complessi
antigene-anticorpo solubili, con moderato eccesso di antigene.
Riguardano specialmente antibiotici come il cefaclor (ca. 1% dei
bambini), ma anche altri farmaci: l'uso di anticorpi monoclonali ha
riportato d'attualità la malattia da siero. La SSLR si
manifesta con rash orticarioide, artralgia/artrite e spesso febbre.
La proteinuria può essere presente e nella paziente n. 10 ha
costituito il segno fortuito di esordio. Quasi mai si trovano livelli
elevati di immunocomplessi, poiché questi sono presenti in
fase preclinica. Si conferma la maggior frequenza di reazioni a
farmaci nelle femmine (9 pazienti su 11). Il periodo di latenza
dopo un primo contatto col farmaco (generalmente 7-12 giorni nella
forma classica) è il tempo necessario a formare una
sufficiente quantità di anticorpi. Se l'organismo era già
stato trattato in precedenza con lo stesso farmaco o un altro
cross-reattivo (v. caso 3), la risposta anticorpale non è più
primaria ma secondaria e quindi più rapida oltre che più
intensa (forma accelerata). La presenza di casi atipici - nel caso 10
l'intervallo di latenza è rimasto invariato, nel 2 si è
progressivamente allungato - fa supporre che i meccanismi della SSLR
possano essere più complessi. Il caso 2 pone interrogativi
sulla possibilità che, almeno in certi casi, la risposta
allergica all'antibiotico possa essere seguita da tolleranza:
durante il TPO con amoxicillina, infatti, la bambina ha mostrato
alcuni segni di laboratorio di reazione tipo III senza alcuna
manifestazione clinica come se il suo organismo fosse arrivato a
tollerare l'antibiotico.
Clinica
Pediatrica, IRCCS pediatrico Burlo Garofolo, Trieste
Il
registro italiano delle CAPS (Cryopyrin-Associated Periodic Syndrome)
è iniziato nel 2004 e attualmente ha arruolato 29 pazienti di
età media di 19,6 anni. Si tratta di: 16 pazienti con sindrome
CINCA (9 maschi e 7 femmine), 8 pazienti con sindrome di MW (4 maschi
e 4 femmine) e 5 pazienti affetti da FCU (2 maschi e 3 femmine). Dal
2005 abbiamo iniziato a trattare questi pazienti anakinra
(antagonista del recettore dell'IL-1) alla dose di 1 mg/kg/die: i
16 pazienti trattati hanno mostrato un rapido miglioramento sia
clinico che laboratoristico. I restanti 6 non trattati hanno
presentato una continua evoluzione della malattia (Lepore L. et al. J
Pediatr 2010) registro. a oggi, 8 dei 16 pazienti in trattamento
iniziale con anakinra, sono passati al trattamento con canakinumab
(anticorpo monoclonale umano anti IL-1?) alla dose iniziale di 150
mg ogni 8 settimane. Tutti hanno mantenuto la costante remissione di
malattia. Di questi, 5 hanno richiesto un aumento della dose a 300 mg
dì. Dei pazienti che hanno mantenuto la terapia con anakinra,
tutti tranne due sono rimasti in remissione. In un paziente si è
assistito a una ripresa del rash e comparsa di artrite, giustificata
dalla scarsa aderenza alla terapia (terapia occasionale e
discontinuativa con anakinra); un secondo paziente ha ripresentato il
rash cutaneo malgrado la terapia adeguata. Diversamente il dimorfismo
(facies tipica) e le alterazioni ossee sono rimaste costantemente
presenti, senza però ulteriori peggioramenti, così come
la sordità e il papilledema. Non sono stati osservati effetti
avversi nei pazienti in terapia con anakinra. Si segnalano invece due
effetti collaterali secondari alla somministrazione di canakinumab: 1
paziente ha presentato vertigine e 1 paziente ha manifestato rialzo
pressorio. Dei 6 pazienti inizialmente non trattati (per rifiuto
della famiglia, o malattia lieve all'esordio) 3 hanno iniziato
terapia con canakinumab, 1 è stato perso al follow-up e 2
restano ancora non trattati. Questi ultimi hanno mostrato un decorso
progressivamente evolutivo della malattia con la comparsa di nuove
manifestazioni cliniche e indici di flogosi sempre elevati. Per
quanto riguarda i pazienti trattati, tutti hanno risposto alla
terapia e negativizzato gli indici di flogosi. Non si segnalano
effetti collaterali secondari alla terapia. A questi, nell'ultimo
anno, si sono aggiunti 8 nuovi pazienti (5 FCU e 3 MWS). Di questi, 2
pazienti con sindrome di MWS hanno iniziato terapia con anakinra alla
dose di 1 mg/kg/die con ottima risposta alla terapia (scomparsa di
febbre, rash, artrite, artralgie). Uno dei due dopo un periodo di
trattamento di 9 mesi, ha avviato terapia con canakinumab mantenendo
la costante remissione di malattia.In conclusione il registro
italiano ha permesso di raccogliere nel tempo un numero sempre
maggiore di casi di CAPS, di seguire la loro storia naturale dando a
tutti i pazienti l'opportunità di essere trattati con i
farmaci inibitori di IL-1. Tali farmaci hanno cambiato la storia
naturale di queste rare condizioni, bloccando l'evoluzione della
malattia e impedendo le sue gravi complicanze. Il lungo follow-up dei
pazienti trattati con anakinra ha potuto stabile oltre all'efficacia,
la sua buona tollerabilità. La stessa valutazione sarà
fatta per canakinumab che appare altrettanto efficace ma i cui
effetti collaterali andranno verificati nel tempo. Per ora possiamo
affermare che la modalità di somministrazione di canakinumab
(sottocute ogni 8 settimane) è un sicuro vantaggio, specie in
pediatria, mentre la lunga emivita del farmaco impone una stretta
sorveglianza in caso di infezioni.
Pediatria
OCL, Lugano (Svizzera)
Introduzione.
La nefrite lobare acuta (ALP) è un'infezione renale acuta
severa localizzata. Appare come una massa infiammatoria senza segni
di liquefazione, coinvolgente uno o più lobi o lobuli renali.
Caso
clinico. Ragazzina lamenta febbre, cefalea frontale e
addominalgia al fianco dx, pollachiuria e disuria. Sofferente,
dolorabilità in fianco e fossa iliaca dx con difesa, Giordano
positivo, rialzo di indici di flogosi e leucocituria. Nel sospetto di
pielonefrite, sebbene sonografia negativa, inizia terapia con
ceftriaxone 55 mg/kg/die ev. Dopo 24 h, aggravamento e, con clinica e
controllo sonografico suggestivi, viene eseguita appendicectomia,
anatomopatologicamente indenne. Dopo fugace miglioramento clinico,
ennesimo peggioramento con rialzo indici di flogosi, ecocardiografia
e Rx torace nella norma, ma TAC addominale diagnostica per nefrite
lobare dx in presenza di un doppio distretto ureterale. Trattata con
ceftriaxone ev 100 mg/kg/die per 21 gg, beneficio clinico e
apiressia.
Discussione.
L'ALP presenta febbre, dolore al fianco, piuria, leucocitosi,
batteriuria, ma emocolture e urinocolture possono risultare negative.
La diagnosi si avvale di immagini sonografiche associate a TC, dove
si differenziano due tipi di lesioni, semplici o complesse, con
caratteristiche cliniche e morfologiche differenti. Non vi è
consensus sulla durata della terapia, recenti studi propongono
trattamento parenterale per 3 settimane con buoni risultati clinici.
Vi è un rischio maggiore di scar renali nelle ALP rispetto a
pielonefriti.
Conclusioni.
Il caso presentato dimostra come l'ALP, infezione renale severa non
comune, possa non essere facilmente diagnosticata (urinocolture ed
emocolture negative, clinica mima appendicopatia) e come si possa
drammaticamente risolvere con terapia adeguata e prolungata.
Clinica
Pediatria, IRCSS pediatrico Burlo Garofolo, Università
di Trieste.
Vediamo
M., 13 anni, al terzo episodio febbrile nell'arco di due mesi. Il
quadro si era associato in ogni occasione a indici di flogosi elevati
(PCR nell'ordine di 10 mg/dl), ma per il resto la sintomatologia
era sempre stata aspecifica (stato confusionale, vomito) e le
indagini radiologiche non avevano permesso di evidenziare un focus
infettivo.
La sua
storia era iniziata, in particolare, con un episodio di febbre
elevata associato a stato confusionale e crisi di agitazione.
Ipotizzando un quadro neurologico erano state eseguite TAC, RMN
cerebrali, EEG ed esame del liquor, risultate negative. Nel sospetto,
seppur dubbio, di un'encefalite era stata avviata una terapia
empirica con aciclovir e ceftriaxone ev, con successiva miglioramento
clinico. Dopo due settimane di benessere tuttavia M. ripresentava
picchi febbrili associati a vomito e stato lievemente soporoso.
L'emocultura e la RMN encefalo risultavano ancora una volta
negative e anche la radiografia del torace, l'ecocardiografia,
l'ecografia addominale e l'esame delle urine non evidenziavano
quadri patologici. Il quadro clinico rispondeva nuovamente al
trattamento empirico con ceftriaxone e M. veniva dimesso dopo 5
giorni di terapia ev. Poco tempo dopo la sospensione degli
antibiotici tuttavia compariva nuovamete la febbre, associata a
vomito e lieve dolore addominale. Risultando ancora una volta
negative le indagini di primo livello (RX torace, eco addome, esame
urine), nel sospetto di un'infezione endoaddominale profonda veniva
eseguita una TAC addominale, che permetteva di evidenziare un'area
di disomogeneità di 5 mm al polo superiore del rene destro,
con piccola area colliquativa al suo interno, in un quadro suggestivo
di ascesso renale. La successiva terapia antibiotica con tobramicina
e teicoplanina ev (per 3 settimane), seguita da terapia orale con
amoxicillina+ acido clavulanico (per ulteriori 3 settimane), portava
poi ad una risoluzione definitiva del quadro.
L'ascesso
renale rappresenta un'evenienza rara in pediatria, con frequente
ritardo di diagnosi a causa del basso indice di sospetto. I sintomi
di accompagnamento non sono sempre specifici e la febbre può
rappresentare l'unico sintomo di rilievo. L'esame urine può
essere negativo e la sensibilità dell'ecografia non è
assoluta: nei casi sospetti è dunque raccomandata l'esecuzione
di una TAC addominale.
F.M.
Bosetti, M. Bianciotto, G. Migliore, R. Pagliero, A.F. Urbino
Pediatria
D'Urgenza, Ospedale Infantile Regina Margherita, Torino
L'ascesso
renale è una patologia poco comune in età
pediatrica1,2. I patogeni (stafilococchi nell'80% dei
casi, e in minor misura Gram-negativi) possono diffondere per via
urinaria ascendente, ematogena o per continuità2.
La sintomatologia è aspecifica1 e spesso i colturali non sono
dirimenti, perciò la conferma diagnostica si basa sull'imaging
e indirettamente sulla mancata risposta alla terapia antibiotica1,2.
Descriviamo
il caso di un bambino di 6 anni, ricoverato per febbre da 5 giorni,
in malessere e con netto incremento degli indici di flogosi.
Obiettività negativa salvo numerose carie destruenti.
Nonostante la terapia antibiotica, persistevano febbre ed elevati
indici di flogosi con colturali negativi. Per la comparsa di dolore
addominale si eseguivano Rx addome diretto (negativa) ed ecografia
addominale indicativa di ascesso renale sinistro. Veniva quindi
sospeso ceftriaxone e introdotto meropenem con rapido sfebbramento.
La diagnosi veniva confermata con TC e RM addome. La necessità
di una tempestiva terapia antibiotica non ha consentito di isolare il
patogeno ma un'accurata valutazione odontoiatrica poneva l'ipotesi
di una causa odontogena del processo infettivo.
La
diagnosi precoce è essenziale per minimizzare il danno
parenchimale2, pertanto l'ipotesi di ascesso renale va presa in
considerazione in pazienti che presentino febbre, dolore addominale e
netto incremento degli indici di flogosi, in particolare in presenza
di un quadro suggestivo di IVU refrattaria a terapia antibiotica
adeguata3.
Bibliografia
- Wang YT, et al. Renal abscess in children: a clinical retrospective study. Acta Paediatr Taiwan 2003;44:197-201.
- Angel C, et al. Renal and peri-renal abscesses in children: proposed physio-pathologic mechanism and treatment algoritm. Pediatr Surg Int 2003;19:35-9.
- Fullà J, et al. Abscesos renales y perirenales: analisis de 44 casos. Rev Chil Ingect 2009;26:445-51.
U.O.
Conegliano (Treviso); Scuola di Specializzazione in Pediatria,
Università di Trieste
G. è
un bimbo di 5 anni. La sua storia inizia una domenica pomeriggio
quando, a una sagra con la nonna, inizia a lamentare un forte mal
di pancia, tanto da non voler camminare. La nonna interpreta il
dolore come dovuto a un fecaloma (il bimbo infatti è stitico),
comunque a casa il mal di pancia cessa.
Lunedì
G. scarica, sta bene, però la sera presenta una puntata
febbrile (T 38,5 °C) a ciel sereno, per cui la nonna gli
somministra del paracetamolo.
Il giorno
successivo sta nuovamente bene, gioca e corre all'asilo, ma la sera
dice di sentire tanto freddo e ha i brividi: la nonna riscontra una
temperatura di 39 °C, e somministra paracetamolo. Il bimbo dorme
qualche oretta, ma nel cuore della notte si sveglia lamentando un
fortissimo dolore addominale, localizzato soprattutto al fianco e
fossa iliaca destra. La nonna, pensando ancora alla stipsi, gli fa un
clistere, G. scarica, ma il dolore continua. Chiama così la
Guardia Medica che, nell'ipotesi di un'appendicite, manda il
bimbo in ospedale.
All'ingresso
in PS, G. è sofferente, piange per il dolore, T 39,4 °C.
Lo visitiamo. L'addome è trattabile, ma il bimbo continua a
lamentare un forte dolore in fossa iliaca e fianco dx. Al torace si
riesce ad ascoltare qualche piccolo (e dubbio) crepitio alla base. Lo
stix urine mostra solo la presenza di chetoni (++). All'emocromo,
leucocitosi (18.000 GB), con indici di flogosi negativi (PCR 0,2).
Gli somministriamo del paracetamolo e lo teniamo in osservazione. La
mattina successiva, G. si sveglia tranquillo, apiretico, ma ha ancora
mal di pancia, questa volta diffuso, con obiettività
addominale negativa. Anche il torace è negativo. È
comparso però qualche accesso di tosse catarrale. Nell'ipotesi
di polmonite, eseguiamo una Rx torace, che conferma il nostro
sospetto.
Conclusioni:
In corso di polmonite a interessamento basale è frequente la
presenza di un dolore addominale riferito, che può talora
simulare un'appendicite. Non lasciamoci ingannare, tanto più
se l'obiettività addominale è negativa.
Un
virus a gambe larghe
Clinica
Pediatrica, IRCCS pediatrico Burlo Garofolo, Trieste
T. è
un bambino di 2 anni giunto alla nostra osservazione, trasferito da
un'altra Struttura per insorgenza acuta di disturbi dell'equilibrio
e difficoltà alla deambulazione, insorti dopo 3 giorni di
diarrea acquosa ed episodi di vomito. Esami ematici, visita
oculistica ed ORL risultavano nella norma. Il bambino veniva
trasferito presso il nostro reparto per eseguire esami di
approfondimento diagnostico. Anamnesi negativa. Vaccinazioni di legge
e facoltative eseguite All'esame obiettivo, Tommaso si presentava
in condizioni generali discrete, idratazione ai limiti inferiori e
refill capillare < 2"; andatura atassica, incapacità a
mantenere la stazione eretta. Non deficit dei nervi cranici né
di forza. Buon controllo del capo. Pupille isocoriche isocicliche,
normoreagenti allo stimolo luminoso. Ammiccamenti palpebrali
bilaterali. TC 37.7 °C. Posizionata fleboclisi ed iniziata
reidratazione ev. Venivano eseguite TAC cranio, che escludeva segni
di ipertensione ed emorragie endocraniche, e rachicentesi (esami
chimico del liquor e colturale negativi). L'EEG eseguito in fase
acuta e la RMN cranio-midollo risultavano negativi. Gli esami
microbiologici (tamponi, sierologie, coprocolture) sono risultati
negativi, eccetto per la presenza del Rotavirus nelle feci. Il
Rotavirus è uno dei principali patogeni causa di
gastroenterite in età pediatrica; manifestazioni neurologiche
si verificano approssimativamente nel 2-5% dei pazienti con
sintomatologia gastrointestinale. In Letteratura sono riportati casi
isolati di atassia da rotavirus; peraltro, alcuni dati confermano un
certo grado di neurotropismo di questo virus, oltre che il possibile
riscontro del virus o di tracce del suo genoma nel liquor in pazienti
con gastroenterite e atassia (ma anche la sua occasionale presenza
nel liquor in corso di gastroenterite in assenza di manifestazioni
neurologiche), la segnalazione di una fase viremica nei primi giorni
dall'esordio della gastroenterite da rotavirus e la documentazione
di una tendenza invasiva extraintestinale da parte di alcuni ceppi.
Non è chiaro come il Rotavirus possa colpire il SNC anche
senza un'invasione diretta; tuttavia da recenti studi risulta
ipotizzabile che il danno possa essere di natura infiammatoria
secondario all'azione di interleuchine, attivate dal rilascio di
una tossina di origine virale.
UO
Pediatria 1, Azienda Ospedaliero-Universitaria Pisana
Caso
Clinico. A., 19 mesi, è stato condotto alla nostra
osservazione per la presenza di una tumefazione in regione parietale
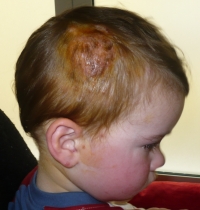
destra di forma rotondeggiante (diametro massimo di circa 5 cm) con
margini irregolari (Figura 1). La lesione
appariva iperemica e crostosa, e presentava gemizio di materiale
purulento; in tale sede i capelli apparivano radi e facilmente
asportabili. Alla palpazione la lesione risultava dolorabile e di
consistenza teso-elastica. A livello del padiglione auricolare e in
regione retro-auricolare destra e alla base del collo erano evidenti
alcune lesioni satelliti di piccole dimensioni (mm). Il paziente
presentava inoltre linfoadenomegalia retro-auricolare,
sotto-angolo-mandibilare e latero-cervicale omolaterale, ed era in
buone condizioni generali e apiretico.
Gli esami
ematici mostravano una modesta positività degli indici di
flogosi (PCR 1,24 mg/dl, v.n. < 0,5; PCT 0,03 ng/ml, v.n. <0,05)
e leucocitosi (GB 22.330/mm3; N 45,8%).
Dall'anamnesi
emergeva che circa 20 giorni prima il bambino aveva iniziato a
presentare in sede parietale destra una lesione eritematosa nummulare
(diametro di circa 1 cm), non pruriginosa, finemente desquamante.
Tale lesione si associava a linfoadenomegalia retro-auricolare
omolaterale. Nei giorni successivi la lesione era aumentata di
dimensioni, con notevole incremento della componente flogistica e con
la comparsa di gemizio spontaneo di materiale purulento e
dolorabilità alla digitopressione. Per tale motivo il paziente
era stato sottoposto a terapia antibiotica topica con
gentamicina-betametasone e con amoxicillina-acido clavulanico per via
sistemica, senza nessun miglioramento.
Durante
la valutazione del paziente in PS (dove i genitori si erano rivolti
per il mancato miglioramento della lesione), nel sospetto di una
raccolta ascessuale il bambino è stato sottoposto a
visita chirurgica nell'eventualità di un possibile drenaggio
della stessa. Un esame ecografico della tumefazione, a sorpresa,
metteva in evidenza esclusivamente un ispessimento del tessuto
superficiale sottocutaneo, senza visualizzabili raccolte ascessuali.
Il bambino è stato quindi inviato in Pediatria per ulteriori
accertamenti. All'esame clinico veniva posto il sospetto di Kerion
Celsi da tinea capitis, per cui il bambino è stato
sottoposto a visita dermatologica che confermava la diagnosi.
Il
bambino è stato sottoposto a terapia antimicotica sistemica
con griseofulvina (20 mg/kg/die per os x 2 volte/die per 8 settimane)
e topica con tioconazolo (dopo rasatura dei capelli, 2 volte/die per
30 giorni). Gli esami colturali eseguiti hanno successivamente
confermato la presenza di miceti, ma non è stato possibile
identificare la specie in causa. Tuttavia, poiché il bambino
viveva in una zona rurale con numerosi animali domestici (cani) era
ipotizzabile un'infezione micotica da dermatofita zoofilo.
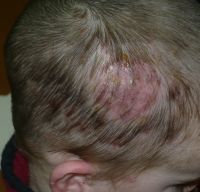
Alla
visita di controllo, eseguita dopo 7 giorni di terapia, la
tumefazione risultava notevolmente ridotta di dimensioni, con
riduzione dei caratteri flogistici e quasi completa scomparsa delle
lesioni pustolose (Figura 2). Nonostante la
buona risposta terapeutica, il ritardo nella diagnosi esiterà,
verosimilmente, con la presenza di un'area di alopecia cicatriziale
nella sede della lesione.
Conclusioni
⢠Il
Kerion Celsi è espressione di una marcata reazione flogistica
nei confronti di una tinea capitis causata da dermatofiti zoofili.
⢠L'aspetto
simil-ascessuale della lesione, associato alla marcata flogosi
locale, può indurre facilmente in un errore diagnostico e
terapeutico che potrebbero condizionare la restitutio a integrum.
⢠In
presenza di una lesione cutanea dall'eziologia incerta è
consigliabile non intraprendere un trattamento cortisonico topico.
⢠La
diagnosi di infezione fungina e il trattamento antimicotico devono
essere precoci per ridurre il rischio di esiti permanenti come
l'alopecia cicatriziale.
 |
 |
Bibliografia
di riferimento
- Elewski
BE. Cutaneous mycoses in children. J Am Acad Dermatol 2000;42:1-20.
- Hackett
BC, et al. Tinea capitis in a paediatric
population. Ir Med J 2006;99:294-5.
- Mazzatenta
C. Kerion Celsi. Medico e Bambino 2007;9:593-4.
- Pomeranz
AJ, Sabnis SS. Tinea capitis: epidemiology, diagnosis and management
strategies. Pediatr Drugs 2002;4:779-83.
Dipartimento
dell'Età Evolutiva, Clinica Pediatrica, Università
degli Studi di Parma
Una
bambina di origine indiana di 10 anni e 4 mesi è giunta alla
nostra osservazione per episodio caratterizzato da cefalea frontale,
pallore cutaneo con successiva perdita di coscienza e scosse
generalizzate, con movimenti afinalistici della lingua, della durata
di circa 2-3 minuti, seguito da stato post-critico e da due episodi
di vomito. Non riferiti febbre né altri disturbi nei giorni
precedenti. Anamnesi fisiologica non degna di nota. Anamnesi
familiare muta per crisi comiziali. Anamnesi patologica remota
negativa per episodi analoghi; non riferiti pregressi traumi cranici.
All'ingresso obiettività clinica e neurologica nei limiti
della norma. Eseguiti ECG, esami ematochimici e misurazione della
pressione arteriosa, risultati nella norma. Dato il recente
trasferimento dal paese di origine sono state inoltre eseguite
coprocolture per batteri e parassiti risultate negative. è
stata eseguita anche consulenza NPI ed elettroencefalogramma che ha
mostrato la presenza di eccesso di attività lenta focale in
ambito emisferico sinistro, frammista ad attività sporadica.
Durante la degenza la bambina si è sempre mantenuta in buone
condizioni cliniche e non ha presentato ulteriori episodi comiziali.
Eseguita RMN encefalo con riscontro di lesione intra-assiale
iuxta-corticale, parietale sinistra, rotondeggiante, disomogenea con
caratteristiche di segnale a bersaglio, del diametro massimo di
circa 9,5 mm, circondata da estesa quota di edema perilesionale; si
associa appianamento degli spazi subaracnoidei. Tale lesione mostra
relativa iperintensità centrale e presenta, dopo
somministrazione di mdc, enhancement a cercine periferico
sottile e di spessore regolare. I rilievi descritti appaiono
riferibili a lesione focale a verosimile eziologia
infettivo-infiammatoria (tubercoloma? lesione micotico-parassitaria
tipo cisticercosi?). Si decide di eseguire accertamenti eziologici in
relazione alla lesione parietale sinistra e di intraprendere terapia
anti-edemigena con mannitolo ev in relazione al reperto di edema
perilesionale. Le caratteristiche della lesione neuroradiologica
hanno indotto ad eseguire accertamenti diagnostici differenziali tra
neurocisticercosi e lesione tubercolare; pertanto sono state eseguite
indagini infettivologiche (Mantoux, Quantiferon-TB Gold, ricerca BK
nell'escreato e nell'urina) e radiologiche (Rx torace) che hanno
permesso di escludere una neuro-TBC. Sono stati inoltre richiesti
marker sierologici di immunodeficienza acquisita e di epatite virale
risultati negativi. Eseguite anche ulteriori indagini
parassitologiche su siero per la ricerca di anticorpi
anti-echinococco, anti-toxocara canis e anti-tenia solium risultati
negativi. Nonostante la negatività degli anticorpi anti-Tenia
solium, data la presenza di lesione cerebrale altamente suggestiva,
della provenienza da area endemica e sulla base dei criteri
diagnostici attualmente approvati, si è convenuto per una
diagnosi di probabile neurocisticercosi. è stata pertanto
intrapresa terapia con Albendazolo e Desametasone. Nell'ipotesi
diagnostica di neurocisticercosi sono state inoltre eseguite visita
oculistica, ecografia addominale e consulenza dermatologica che hanno
permesso di escludere la disseminazione di eventuali cisti ad altri
organi. La bambina è stata dimessa, consigliando di proseguire
a domicilio terapia con Albendazolo per un totale di 19 giorni. Nel
mondo si stima che ci siano 50-100 milioni di persone infestate da T.
solium. Le aree di maggiore endemia comprendono America Latina,
India, Cina, Sud Est Asiatico e Africa sub-Sahariana. In America
Latina ed in India la sieroprevalenza per teniasi varia dal 5-25%. La
prevalenza di crisi epilettiche nei Paesi poveri è doppia
rispetto a quella dei Paesi ricchi. La diagnosi è talvolta
difficile perché le manifestazioni cliniche non sono
specifiche, la maggior parte dei quadri neuroradiologici non sono
patognomonici ed i test sierologici sono poco sensibili e poco
specifici.
Clinica
Pediatrica, IRCCS pediatrico Burlo Garofolo, Trieste
A. è
un bambino di 4 anni che giunge alla nostra attenzione perché
da diversi giorni la mamma ha notato che il piccolo mantiene la testa
inclinata dal lato sinistro e presenta un vomito mattutino
persistente. Nelle ultime tre settimane A. aveva presentato prima
un'otite media acuta trattata, dato il persistere della febbre, con
8 giorni di amoxicillina e poi una gastroenterite severa tanto che
era stata necessaria una terapia idratante endovenosa.
Da allora
le scariche si erano risolte ma persisteva un vomito quotidiano dopo
colazione su cui la mamma era stata tranquillizzata dalla curante
inquadrandolo come coda della gastroenterite. Tuttavia alla mamma A.
sembrava strano, diceva che non era più lui, e alla
comparsa di un lieve strabismo intermittente si è precipitata
in ospedale.
Obiettivamente
il piccolo presentava un'esotropia lieve all'occhio sinistro ma
anche inclinazione del capo verso destra, libero nei movimenti.
Storia di
otite-disidratazione-bambino-che-non-era-più-lui (frase
che ho imparato essere di per sé stessa allarmante). Da qui
all'esecuzione della RMN cerebrale il passo è stato breve,
anche se si è passati attraverso una valutazione del fundus
oculi, che sebbene risultato alterato, non sarebbe stato comunque un
esame conclusivo.
Diagnosi:
trombosi del seno venoso, trasverso e sigmoideo. E mastoidite
radiologica.
L'avvio
della terapia anticoagulante (eparina frazionata prima e dicumarolo
poi) ha portato a una progressiva risoluzione del vomito e un
miglioramento dello strabismo.
Dall'anamnesi
familiare è emerso inoltre che la mamma è portatrice
del fattore V di Leiden. Il piccolo risulta essere portatore
eterozigote della mutazione (R506Q), ma anche portatore eterozigote
per il gene della MTFHR.
Scuola
di specializzazione in Pediatria, Università di Trieste
T. viene
alla nostra attenzione all'età di un anno e nove mesi per
ipotonia diffusa e difficoltà alla deambulazione, possibile
solo con appoggio. Alla visita si rileva una lieve ipotrofia delle
masse muscolari, aracnodattilia, iperlassità legamentosa e un
ritardo dello sviluppo psicomotorio. Il bimbo pur avendo una facies
triangolare con orecchie grandi e a impianto basso, somiglia molto al
padre. In anamnesi è presente una lussazione congenita delle
anche e un'ernia inguinale. È inoltre presente ipercheratosi
a livello degli arti superiori e inferiori. Il laboratorio mostra
valori elevati delle CPK e delle transaminasi. La visita oculistica
esclude un prolasso del cristallino e con esso il sospetto di una
sindrome di Marfan o una sindrome di Ehlers-Danlos. Anche la visita
cardiologica e l'ecografia dell'addome risultano nella norma. In
considerazione del quadro clinico e laboratoristico prettamente di
carattere muscolare, si decide di eseguire una biopsia muscolare,
effettuata alla coscia destra, che esita con la comparsa di cheloide
rosso rilevato. Sulla biopsia viene eseguita l'analisi
immunoistochimica per la ricerca del collagene di tipo VI e della
merosina, che, nel caso di T., risultano meno rappresentati a livello
tissutale rispetto al controllo. Sulla base del referto istologico,
viene successivamente eseguita l'indagine molecolare che evidenzia
un genotipo eterozigote per la mutazione missenso nell'esone 10 del
gene COL6A1 compatibile con miopatia di Bethlem. Si tratta di una
malattia autosomica dominante, la prevalenza è di 0,5:100.000
nati, si manifesta con debolezza muscolare a carico dei distretti
prossimali, contratture in flessione a carico dei distretti distali e
iperlassità legamentosa di mani e piedi. Questo disordine può
esordire nella prima decade di vita con ipotonia e ritardo
psicomotorio. La diagnosi differenziale si pone con la distrofia
muscolare congenita di Ullrich, l'atrofia muscolare spinale,
sindrome di Ehlers-Danlos e sindrome di Marfan. Il distretto cardiaco
e quello oculare non sono interessati, mentre il coinvolgimento dei
muscoli respiratori, sebbene poco frequente, può richiedere
l'ausilio della ventilazione notturna. Tipica è
l'ipercheratosi follicolare e la formazione di cheloidi.
Infezione
post-natale da CMV come trigger di grave espressione clinica di BRIC
tipo 2 in eterozigosi
1Scuola
di specializzazione in Pediatria, Università di Modena e
Reggio Emilia
2UO
Pediatria, Dipartimento Integrato Materno-Infantile, Azienda
Ospedaliero-Universitaria, Policlinico di Modena
PFIC e
BRIC sono epatopatie ereditarie autosomiche recessive con mutazioni
dei geni ATP8B1 (PFIC/BRIC tipo 1), ABCB11 (PFIC/BRIC tipo 2) e ABCB4
(PFIC tipo 3). Le acutizzazioni di BRIC, spesso associate a infezioni
virali intercorrenti, si manifestano con ittero, prurito, elevazione
di sali biliari, bilirubina, transaminasi, ?FP, con normali yGT,
determinando epatite gigantocellulare, autolimitantesi. L'infezione
postnatale da CMV è riscontrabile sierologicamente o
microbiologicamente oltre le prime 2 settimane di vita associata a
esami negativi nelle prime 2 settimane o su sangue secco della
Guthrie card. Nel nato a termine raramente determina conseguenze
cliniche rilevanti, pur esistendo la possibilità di quadri
severi (epatiti, polmoniti, linfoadenopatie), soprattutto nel LBW.
L'epatite si manifesta con epatosplenomegalia, lieve ittero e
aumento moderato delle transaminasi. L'istologia tipica evidenzia
cellule giganti e multinucleate con inclusioni intranucleari da CMV e
pseudolumi con virioni visibili nei nuclei e nel citoplasma.
Solitamente l'infezione acquisita si risolve spontaneamente e il
trattamento va riservato ai quadri clinici severi. Descriviamo il
caso di RA nato a 40 w, PN 2,8 kg, da parto eutocico; regolare
adattamento alla vita extrauterina, allattamento materno esclusivo. A
68 giorni ittero colestatico persistente, epatosplenomegalia,
ipertransaminasemia, forte aumento di ?FP, lieve iper-yGTemia.
All'ecografia addome epatosplenomegalia con areole iperecogene
pseudonodulari. Sierologia negativa. Escluse patologie neoplastiche
(RMN addome, NSE e ?HCG negative), malattie metaboliche, compresa
tirosinemia, m. di Wilson, s. di Alagille, epatiti autoimmuni.
Individuato CMV su urine, confermato da PCR su sangue, inizia terapia
con ganciclovir ev e AUDC. L'infezione congenita è esclusa
da sierologia materna negativa durante l'ultimo trimestre e PCR
negativa su Guthrie card, confermando l'infezione postnatale
trasmessa mediante latte materno (CMV positivo su latte). Segue
peggioramento del quadro con petecchie e alterazione della
coagulazione. Escludiamo malattia da accumulo lisosomiale tramite
aspirato midollare negativo. La biopsia epatica mostra epatite
giganto-cellulare aspecifica in assenza di inclusioni da CMV. Le
indagini genetiche individuano la mutazione 1331T>C (Val 444Ala)
in eterozigosi del gene ABCB11 responsabile di PFIC/BRIC 2. RA è
in buone condizioni generali in assenza di sintomi e alterazioni
biochimiche. L'andamento clinico e bioumorale permette di
ipotizzare che l'infezione postnatale da CMV sia stata il fattore
precipitante la patologia colestatica di base presente in RA in forma
eterozigote. La concomitanza delle due condizioni, che presenti
singolarmente possono evolvere in modo asintomatico/subclinico, ha
determinato un quadro di epatopatia acuta importante a lenta
risoluzione. Un esordio clinico grave e precoce associato a infezione
da CMV non era finora stato descritto in letteratura in pz
eterozigoti in età pediatrica.
U.O.
Neonatologia, Dipartimento Materno-Infantile, AOUP, Pisa
Caso
clinico. D. nasce a termine da genitori senegalesi consanguinei.
A 29 settimane di EG una RMN dell'encefalo fetale aveva evidenziato
un quadro compatibile con emimegalencefalia. In prima giornata di
vita comparsa di episodi parossistici tipo spasmi tonici asimmetrici
in serie. L'EEG, l'ECO e la RMN cerebrali confermano la presenza
di una emimegalencefalia dell'emisfero sx. La neonata inizia
terapia con fenobarbital, ma per la persistenza di episodi
pluriquotidiani di spasmi asimmetrici e crisi focali motorie viene
aggiunta alla terapia vigabatrin e clonazepam. Per la presenza di una
chiazza ipomelanotica al volto, si richiede consulenza dermatologica
che pone il sospetto di ipomelanosi di Ito. A 2 mesi di vita viene
eseguito un intervento di emisferectomia funzionale, che non risolve
le crisi e richiede un successivo intervento chirurgico.
Discussione.
L'emimegalencefalia è un quadro malformativo cerebrale
complesso caratterizzato da profonda alterazione di parte o di un
intero emisfero cerebrale, che si presenta di dimensioni aumentate.
Si distinguono forme isolate e associate a sindrome (sclerosi
tuberosa, ipomelanosi di Ito, sindrome di Sturge-Weber, sindrome di
Proteus). La patogenesi è correlata a vari meccanismi tra cui
alterazioni della differenziazione, della proliferazione e della
migrazione neuronale che intervengono alla 12°-20° settimana
di EG. I bambini affetti presentano fin dalla nascita ritardo
psico-motorio, emiparesi progressiva, e crisi convulsive
farmaco-resistenti. Il sospetto diagnostico può insorgere già
in epoca prenatale grazie a indagini strumentali in gravidanza. La
conferma al momento della nascita si ottiene con l'esecuzione di
EEG e RMN/TC. Il trattamento è mirato al controllo delle crisi
convulsive e prevede un iniziale approccio farmacologico con
fenobarbital, clonazepam, vigabatrin, midazolam, fenitoina. Può
risultare necessario l'intervento chirurgico (emisferectomia o
emisferectomia funzionale o emi-decorticazione). Vi è grande
dibattito su quando si debba ricorrere alla chirurgia: recenti studi
hanno dimostrato che un intervento precoce migliora le crisi e le
performance cognitivo-motorie, mentre non modifica l'evoluzione
dell'emiparesi.
U.O.
Neonatologia, Dipartimento Materno-Infantile, AOUP Pisa
Caso
clinico 1. D. nasce a termine da gravidanza normodecorsa, Apgar
5' = 7 e necessità di supporto respiratorio. In 1°
giornata ipotonia generalizzata, motricità spontanea scarsa,
postura batraciana arti inferiori, facies amimica, scarsa
deglutizione con ristagno salivare, pianto flebile. Anamnesi
familiare muta. Si escludono patologie infettiva, centrale o del 2°
motoneurone, miastenia neonatale, malattie metaboliche e sindromi
genetiche. La biopsia muscolare mostra un quadro compatibile con
miopatia nemalinica. D. va incontro a exitus per complicanza
broncopneumotica grave.
Caso
clinico 2. A. nasce a termine da parto spontaneo, primi atti
fisiologici nella norma eccetto una subito evidente ipotonia
generalizzata con iporeattività, pianto facilmente esauribile,
motricità spontanea assente e fascicolazioni della lingua, ma
meccanica respiratoria e suzione ben conservate. Emocromo, indici
infettivi, screening per malattie metaboliche e Ab anti-recettore Ach
negativi, EMG di dubbia interpretazione. L'estrazione del DNA
mostra assenza degli esoni 7 e 8 del gene SMN1 (telomerico) e assenza
dell'esone 5 del gene NAIP, che permettono la diagnosi di
amiotrofia spinale autosomica recessiva. Exitus a 2 mesi di vita.
Discussione.
L'ipotonia generalizzata è un sintomo neurologico comune in
epoca neonatale; clinicamente il neonato presenta posture anomale,
iperestensibilità delle articolazioni e all'esame obiettivo
positività ai segni della U invertita alla sospensione
ventrale, del pull to sit e della sciarpa. Le cause della
sindrome del floppy infant sono varie. Escluse le cause non
neurologiche (sepsi, ipotiroidismo, patologie del connettivo) è
importante fare una prima distinzione tra danno centrale e
periferico: riflessi presenti o iperevocabili, ipotonia non
paralitica, crisi, letargia e ritardo di sviluppo depongono per una
forma centrale; riflessi assenti o ipoevocabili e ipotonia paralitica
fanno sospettare un disordine periferico. In quest'ultimo caso
risultano fondamentali per la diagnosi l'anamnesi familiare, lo
studio del DNA, l'esecuzione di un'EMG ed eventualmente di una
biopsia muscolare.
Dipartimento
dell'Età Evolutiva, Clinica Pediatrica, Università
degli Studi di Parma
Bambino
nato alla 42ª settimana di gestazione da parto cesareo per
presentazione podalica. Peso alla nascita 3,340 kg, lunghezza 48 cm.
Non problemi perinatali degni di nota. Alla nascita buone condizioni
generali; comparsa di ipotono durante i primi mesi di vita con lieve
ritardo dello sviluppo psicomotorio (controllo del capo a 4 mesi,
stazione seduta a 8 mesi, deambulazione autonoma a 19 mesi, prime
parole a 12 mesi).
Giunge
alla nostra attenzione all'età di 6 anni per scarsa crescita
staturale, difficoltà scolastica e lievi disturbi del
comportamento (iperattività e deficit dell'attenzione).
L'esame
obiettivo evidenzia altezza al 3° percentile, lieve deficit
ponderale, ipertelorismo, ptosi palpebrale sinistra, naso piccolo con
narici anteverse, filtro lungo, impianto basso delle orecchie, ernia
ombelicale, scroto a scialle, lieve lassità legamentosa
distale, brachidattilia e sindattilia II-V dito.
Sono
stati eseguiti Rx mano e polso sx per studio età ossea, con
riscontro di lieve ritardo di maturazione ossea rispetto all'età
cronologica, e cariotipo, risultato normale (XY). Il bambino ha
inoltre effettuato valutazione neuropsichiatria che ha evidenziato
livello cognitivo nei limiti della norma per l'età. Sono
emersi aspetti di distraibilità e difficoltà attentive.
Eseguiti
inoltre ecocardiografia ed ecografia renale risultati nella norma.
Nel
sospetto di sindrome di Aarskog, viene eseguita visita genetica che
ha consigliato analisi molecolare per ricerca di mutazione a carico
del gene FGD1, risultato positivo, confermando il nostro sospetto
diagnostico.
Il
bambino è stato seguito regolarmente presso il nostro centro;
l'altezza si è mantenuta al 3° percentile, ai limiti
inferiori del range familiare, con un normale ritmo di crescita.
Riportiamo
questo caso per sottolineare l'importanza di considerare questa
sindrome nella diagnosi differenziale delle condizioni caratterizzate
da bassa statura e dimorfismi facio-digito-genitali.
La
sindrome di Aarskog, descritta per la prima volta negli anni '70, è
caratterizzata da bassa statura, dimorfismi facciali (ipertelorismo,
cow-lick, rime palpebrali antimongoliche, ptosi palpebrale, naso
piccolo, filtro lungo, narici anteverse), dimorfismi auricolari,
manifestazioni ossee e osteoarticolari, brachidattilia, sindattilia,
solco palmare unico, iperlassità legamentosa, piedi larghi e
piatti, alterazioni genitali (scroto a scialle), criptorchidismo,
ernia inguinale.
La forma
X-linked è associata a mutazione del gene FGD1. La diagnosi è
comunque prevalentemente clinica e si stima che soltanto il 19% dei
soggetti affetti presenti tale mutazione. L'estrema eterogeneità
della mutazione e la frequente presenza di caratteristiche cliniche
sovrapponibili ad altre sindromi rende tale condizione sottostimata e
di difficile diagnosi.
Clinica
Pediatrica, Verona
E., nata
a termine (PN 3350 g). Perinatalità e accrescimento
staturo-ponderale regolari. All'età di 6 mesi la bambina
viene portata in PS (ospedale periferico) per un episodio critico a
risoluzione spontanea caratterizzato da tremori fini peri-orali,
rottura del contatto, ipertono e flessione degli AASS, clonie dell'AS
destro. Gli esami in urgenza mostrano: Na 118 mEq/l, K emolitico, GB
30,600/mm3; EGV pH 7,34, pCO2 40,4, BE -3,5. Viene posta
in infusione per correggere l'iponatriemia (dopo 4 ore Na 128
mEq/l). Nel sospetto di encefalite infettiva viene iniziata terapia
ev con aciclovir, ceftazidime e amikacina. Il tracciato EEG evidenzia
attività lenta theta delta angolare in regione
centro-temporale sinistra continua e di ritmi rapidi a destra. Viene
quindi impostata terapia con fenobarbital. Trasferita presso la
Pediatria del Policlinico GB Rossi (Verona) esegue rachicentesi, RMN
encefalo, fundus oculi, ecografia trans-fontanellare, ECG: tutti gli
accertamenti risultano negativi. Si sospende pertanto la terapia
antibiotica e antivirale. L'esame urine eseguito al momento del
ricovero mostra osmolarità urinaria 42 mOsm/l e PS 1001, con
esami di funzionalità renale, elettroliti e glicemia nella
norma. In seconda giornata di ricovero viene approfondita l'anamnesi
familiare, che rivela una assunzione di liquidi extra-latte pari a
1200-1300 ml/die (PC 8040 g); idratazione 225%. L'ipotesi
diagnostica iniziale viene quindi riconsiderata alla luce dei primi
dati ematochimici e il riscontro iniziale di iponatremia grave con
episodio critico viene collegato a una possibile emodiluizione da
acqua libera. La bimba viene posta in restrizione di liquidi, e il
giorno successivo viene eseguito test di assetamento per escludere la
presenza di una poliuria forzata. Nell'arco delle 4 ore del test
l'osmolarità urinaria aumenta, con minima produzione di
urina e stabilità del peso corporeo. Questo risultato ha
permesso di escludere la presenza di diabete insipido e di formulare
la diagnosi di polidipsia primaria con sindrome da intossicazione
d'acqua.
Clinica
Pediatrica, IRCCS pediatrico Burlo Garofolo, Trieste
B. è
un bambino di 2 anni di origine nigeriana, nato in Italia, che giunge
alla nostra osservazione, trasferito da altra Struttura, dove era
stato condotto per febbre e tosse. L'Rx del torace mostrava un
opacamento del lobo superiore dx e la TC polmonare, eseguita
successivamente, per mancata risposta alla terapia antibiotica,una
massa mediastinica che improntava la trachea. Per il concomitante
riscontro di anemia microcitica sideropenica, nel sospetto di linfoma
era stato eseguito un aspirato midollare (negativo). Era stata
inoltre eseguita la biopsia di un linfonodo laterocervicale dx, che
documentava una flogosi granulomatosa con elementi gigantocellulari.
Le sierologie per HIV ed EBV erano risultate negative.
L'intradermoreazione di Mantoux e il quantiferon erano risultati
francamente positivi nel bambino e nella madre. Era stata dunque
avviata triplice terapia antibiotica antitubercolare e trasferito il
piccolo presso il Nostro Ospedale. All'esame obiettivo si
apprezzavano riduzione del MV, ronchi diffusi e rantoli in sede
medio-basale dx. Inoltre si notavano bozze frontali, solco di
Harrison, varismo agli arti inferiori, braccialetti ai polsi. La
ricerca del MTB nelle urine e negli aspirati risultava negativa. Alla
TC si confermava la massa mediastinica da voluminoso pacchetto
linfonodale colliquato, che comprimeva la trachea e i grossi vasi.
Gli esami ematici mostravano un iperparatiroidismo e ridotti livelli
di 25OHD; l'Rx degli arti inferiori evidenziava lo slargamento
delle mediatisi di tibia e femore. Per la mancata riduzione agli
esami radiologici della massa mediastinica, dopo 4 settimane, si
aggiungeva l'etambutolo e cortisone. Per il rachitismo si
somministrava supplementazione con vitamina D; concomitatamente si
apprendeva la positività dell'esame colturale della biopsia
linfonodale per MTB. La TBC mediastinica è descritta in
Letteratura come estremamente rara in età pediatrica. I
bambini di colore, spesso vitamina D deficienti, sono più
soggetti a contrarre la TBC e ad avere forme più aggressive e
farmacoresistenti di malattia in quanto la Vitamina D è un
potente farmacoregolatore dell'immunità cellulo-mediata. La
supplementazione di vitamina D, in casi come questi, può
permettere una più rapida risposta clinico-radiologica.
Dipartimento
Materno Infantile, Azienda Ospedaliero Universitaria, Policlinico di
Modena
La Shaken
Baby Syndrome (SBS) è il risultato di un violento scuotimento
con o senza contatto con una superficie dura, in cui la vittima è
scossa violentemente, provocando movimenti improvvisi e incontrollati
della testa. Clinicamente la SBS è caratterizzata da segni di
grave o moderato trauma cerebrale con quadri clinici variabili.
Spesso si assiste a una mancata consapevolezza del danno inflitto. I
fattori di rischio sono: pianto inconsolabile; frustrazione e stress
psicofisico dei genitori; basso stato socioeconomico; abuso di droghe
o alcol; gravidanze plurigemellari; personalità borderline con
tendenza al comportamento violento; giovane età della madre;
nascita pretermine; disturbi del sonno e del comportamento del
bambino. La mortalità è del 30%, il deficit neurologico
a lungo termine è presente nel 62-96% dei soggetti. SZ, 3
mesi, ex-prematuro, secondo nato da gravidanza trigemina, giungeva
presso l'Accettazione Pediatrica per episodio di apnea
caratterizzato da ipotono generalizzato, pallore cutaneo al volto,
gemito respiratorio della durata di circa 10 minuti a risoluzione
spontanea. All'esame obiettivo si riscontrava fontanella anteriore
lievemente bombata, in assenza di altra obiettività
patologica. Data la lieve anemia evidenziata dagli esami ematici, si
eseguiva pertanto ecografia cerebrale con riscontro di sanguinamento
extra cerebrale nello spazio sottodurale frontale destro, confermato
dalle indagini neuroradiologiche. All'EEG: onde lente in regione
temporo-occipitale destro. L'RX torace mostrava esiti di frattura
dell'arco medio della V costa di destra. Il FOO evidenziava
emorragie retiniche. Nei 2/3 dei casi con SBS sono presenti emorragie
retiniche, che non correlano mai con trauma cranici di minore entità.
Il controllo del FOO è una manovra minimamente invasiva ma di
grande supporto diagnostico. La SBS deve sempre essere tenuta in
considerazione tra le diagnosi differenziali di emorragia cerebrale e
ALTE (Apparent Life-Threatening Event).
Clinica
Pediatrica, IRCCS pediatrico Burlo Garofolo, Università
di Trieste
F. 9 anni
viene ricoverata per tosse cronica. Tre mesi prima in seguito a
un forte mal di gola, era comparsa una tosse secca e da allora non se
ne era più andata via. Per il problema la bambina aveva perso
molti giorni di scuola. Gli ambiti di patologia organica erano già
stati esclusi con approfonditi accertamenti, ed erano già
state provate senza successo diverse terapie. La tosse si presentava
con delle caratteristiche molto particolari: quotidiana, a orario
(iniziava sempre al pomeriggio verso le 17, e si protraeva fino
all'addormentamento); pur tuttavia la bambina continuava a fare le
varie attività (giocare, mangiare, guardare la televisione),
senza che nulla potesse distrarre la tosse, ma anche senza
essere apparentemente disturbata dal sintomo, se non per il fatto di
faticare ad addormentarsi. Una volta addormentata però la
piccola non tossiva più. Tipicamente si portava la mano
davanti alla bocca.
La
peculiarità è rappresentata dall'aspetto stereotipato
del sintomo, che si manifesta con le medesime specificità e
ricorrenza anche durante il ricovero. Tutte le caratteristiche
descritte erano quelle di una tosse cronica non organica, ma
riferibile a un disturbo di conversione. Il processo di guarigione è
passato attraverso un progetto che ha previsto: 1. la
rassicurazione dei genitori e della bambina sull'esclusione
dell'organicità e sulla benignità del sintomo,
destinato a risolversi, anche se non nell'immediato. 2.
Sull'esplicitare con fermezza la non opportunità a
effettuare ulteriori accertamenti medici. 3. Il contesto
familiare pareva globalmente adeguato, ma emergevano alcune fragilità
nella sfera emotiva di F. che potevano alimentare il disturbo.
Attraverso colloqui di supporto alla genitorialità con una
psicologa sono state fornite specifiche strategie
educative-relazionali. Fondamentale è stato anche sollecitare
la ripresa delle normali attività, sia della famiglia
(monopolizzata dal sintomo), sia della bambina, in particolare della
frequenza scolastica. 4. Infine condivisione del progetto con
il Curante, per una coerenza di messaggi.
Dopo
qualche settimana il disturbo è scomparso.
Scuola
di Specializzazione in Pediatria, Università di Pisa
M. ha
presentato all'età di 4 anni una ipertransaminasemia
asintomatica, di riscontro occasionale con normali livelli circolanti
di gamma-GT (?GT) scoperta in occasione di un ricovero in ospedale
per una gastroenterite da rotavirus. I livelli sierici di
transaminasi e ?GT, ripetuti più volte, si sono mantenuti
moderatamente elevati in assenza di un'evidenza clinica di
epatopatia. Un bilancio diagnostico ha evidenziato una positività
a titolo significativo per autoanticorpi anti-LKM1, che identifica i
pazienti con epatite autoimmune di tipo 2. M. è stata quindi
inserita in un programma di sorveglianza clinica e bioumorale con
controlli trimestrali. La spontanea riduzione delle aminotransferasi
fino alla loro normalizzazione, nonostante la persistenza della
positività autoanticorpale, e la successiva minima
fluttuazione dell'attività aminotransferasica (entro il
doppio dei valori normali), in presenza di un quadro clinico normale,
hanno indotto a procrastinare l'accertamento bioptico.
All'età
di 6 anni (febbraio 2011) la bambina ha presentato un episodio di
faringodinia associato a febbre seguito, dopo pochi giorni, da
poliuria, polidipsia e polifagia. M. è stata quindi ricoverata
presso la nostra Sezione di Endocrinologia e Diabetologia Pediatrica
dove è giunta in buone condizioni generali, senza segni
clinici di evidente chetoacidosi. Agli esami ematochimici la glicemia
è risultata di 351 mg/dl e fatta eccezione per una lieve
ipopotassiemia, tutti gli altri parametri sono apparsi nella norma,
inclusa l'emogasanalisi e le transaminasi. La positività
degli autoanticorpi anti GAD65 (92,84 U/ml, v.n. < 0,90) ha
dimostrato che M. ha sviluppato una condizione di diabete mellito
tipo 1 (DMT1).
L'epatite
autoimmune è una malattia infiammatoria del fegato la cui
prevalenza è di 0,5-1,0/100.000, con frequenza maggiore nel
sesso femminile. L'etiologia è sconosciuta ma si ipotizza
che, nel determinare la malattia, fattori scatenanti (farmaci,
infezioni virali) agiscano su un substrato immunogenetico (aplotipi
HLA DR3-DR4). Ne sono conosciuti due tipi: il tipo 1 caratterizzato
dalla presenza di autoanticorpi SMA (smooth muscle antibodies) e ANA
(anti-nuclear antibodies) e il tipo 2 caratterizzato dalla presenza
di autoanticorpi anti LKM-1 (liver-kidney microsomal-1) e
anti-citosol epatico (LC1). Il tipo 1 rappresenta i 2/3 dei casi e
colpisce sia bambini sia adulti; il tipo 2 è descritto
soprattutto nei bambini. La severità delle due forme è
simile ed entrambe si associano frequentemente ad altre patologie
autoimmuni (20% dei casi) come tiroidite autoimmune, MICI,
vitiligine, DMT1 e sindrome nefrosica.
Dal punto
di vista clinico il 40% dei pazienti presenta una forma
indistinguibile da un'epatite acuta virale; il 25-40% ha un esordio
insidioso con astenia, ittero, cefalea, perdita di peso; in questi
casi la diagnosi viene posta generalmente dopo alcuni mesi. Il 10%
dei pazienti giunge alla diagnosi direttamente per le complicanze
quali ipertensione portale, ematemesi da rottura di varici esofagee,
diatesi emorragica. La diagnosi di epatite autoimmune si basa sulla
presenza di 4 criteri: 1. aumento delle transaminasi sieriche
in assenza di altre malattie epatiche; 2. presenza di
autoanticorpi (SMA/ANA nel tipo 1, LKM-1 nel tipo 2); 3.
aumento delle ?-globuline sieriche; 4. biopsia epatica
suggestiva di epatite. La terapia consiste nella somministrazione di
prednisolone (2 mg/kg/die) in associazione con azatioprina, nei casi
resistenti. L'epatite autoimmune è solitamente responsiva
all'immunosoppressione, ma il 10% dei pazienti sviluppa cirrosi ed
è candidato al trapianto di fegato a distanza di 10-15 anni
dalla diagnosi.
Per
quanto riguarda M., è necessario sottolineare che il suo
aplotipo HLA non è risultato tra quelli a elevato rischio di
sviluppare DMT1, essendo eterozigote per la presenza di un allele
moderatamente associato, come il DRB1 01 (DR1) e dell'altro
negativamente associato con tale condizione, come il DRB1 07:01
(DR7). Questo assetto HLA potrebbe verosimilmente essere in relazione
con l'andamento, almeno per il momento, non evolutivo nel senso di
una chiara epatite autoimmune. A proposito del DMT1, attualmente M.
sta assumendo insulina con basso dosaggio, probabilmente in rapporto
alla presenza di un'attività beta cellulare residua. Sarà
interessante osservare nel tempo l'evoluzione del danno
beta-cellulare.
1UO
Pediatria, Dipartimento Ostetrico Ginecologico e Pediatrico, Az. Osp.
Santa Maria Nuova, Reggio Emilia
2Scuola
di Specializzazione in Pediatria, Università di Modena e
Reggio Emilia
K.,
femmina, 33 mesi, giunta a ricovero per presenza dal giorno prima del
ricovero di lesioni purpuriche, esordite dapprima come rash
maculo-papulare non pruriginoso a carattere simil-orticariode,
successivamente evolute in elementi a carattere vasculitico. Nei
giorni precedenti segnalati frequenti episodi di flogosi delle alte
vie aeree con tosse persistente. In anamnesi cardiopatia congenita
cianogena con ventricolo destro a doppia uscita e ampio difetto del
setto interventricolare; in epoca neonatale intervento palliativo di
bendaggio dell'arteria polmonare per questo in terapia con acido
acetilsalicilico e captopril.
All'ingresso
in Reparto K. si presenta in buone condizioni generali, SaO2:
80% in aa (valori adeguati alla sua cardiopatia di base), FC 126 bpm,
PA: 98/70 mmHg, apiretica. Elementi purpurici rilevati, con aspetto
figurato a coccarda, a livello di guancia sinistra (Figura),
arti superiori e coscia destra, del diametro massimo di 4 cm, non
dolenti alla digitopressione; faringe iperemico, senza essudato e
candidosi al cavo orale; soffio olosistolico 3-4/VI; restante
obiettività clinica generale nella norma, in particolare
assenza di artralgie. Gli esami ematochimici in prima giornata di
ricovero hanno evidenziato eritrocitosi (GR: 6,28 milioni/mm3) con
lieve piastrinopenia (PLT: 87 x 1000/mm3), elevazione del
D-dimero (2109 ng/ml), senza alterazione del restante profilo
coagulativo, incremento dei valori delle CPK (310 U/l), negatività
degli indici di flogosi, normalità della funzionalità
epato-renale e dello esame urine. L'ecocardio di controllo è
risultato sovrapponibile ai precedenti.
La
normalità degli indici di flogosi,dell'assetto coagulativo,
la negatività dell'emocoltura, associati alle buone
condizioni generali della piccola paziente hanno permesso di
escludere sia una malattia emorragica, che una sepsi. Nel sospetto di
vasculite autoimmune sono state avviate indagini di secondo livello
volte a definirne l'eziologia. Nel frattempo K ha iniziato terapia
corticosteroidea e antistaminica per os. Gli esami di laboratorio di
controllo hanno confermato un quadro di eritrocitosi con
piastrinopenia lieve (condizione pre-esistente al ricovero, e
riconducibile alla cardiopatia congenita operata) e documentato una
normalizzazione degli indici di citolisi muscolare. Il complemento,
gli autoanticorpi (ANA, c-pANCA, ENA, anticoagulante di tipo lupico),
l'assetto marziale e proteico sono risultati nella norma. Il
tampone faringeo rapido e la negatività degli anticorpi anti
DNasi B streptococcica hanno escluso un'infezione da SBEGA. L'esame
urine è sempre risultato negativo per proteinuria ed
ematuria;assente la ricerca di sangue occulto fecale. Le indagini
microbiologiche hanno mostrato: positività delle IgM
anti-Mycoplasma pneumoniae, con IgG negative. Negativa la restante
sierologia per CMV, EBV, Toxoplasma, rosolia. La consulenza
oculistica ha escluso interessamento vasculitico oculare.
Durante
la degenza la pz. si è sempre mantenuta apiretica, in ottime
condizioni generali, libera da qualsiasi sintomatologia, con
parametri vitali nella norma. Il quadro cutaneo si è esteso
con comparsa di nuovi elementi a medaglione interessanti i 4 arti,
inizialmente a carattere emorragico, poi virato al brunastro. La
clinica, caratterizzata da esclusivo interessamento cutaneo, insieme
agli esiti degli accertamenti eseguiti, depone per la diagnosi di
edema emorragico acuto dell'infanzia (EEAI).
L'EEAI
è una vasculite leucocitoclastica dei piccoli vasi. Interessa
più spesso bambini di sesso maschile, l'età
d'insorgenza è compresa tra i 2 e i 60 mesi. Si manifesta
clinicamente con petecchie ed ecchimosi con aspetto a coccarda che
divengono successivamente edematose, il volto e le estremità
distali sono siti cutanei elettivi di insorgenza, mentre raro è
l'interessamento viscerale. La diagnosi differenziale dell'AHEI
si pone principalmente con la meningococcemia, sepsi e/o porpora
fulminante, la dermatite neutrofilia febbrile, l'eritema
multiforme, l'abuso, la malattia di Kawasaki e la porpora di
Schönlein-Henoch, Si tratta di una vasculite mediata da
immuno-complessi, che può essere scatenata da pregresse
infezioni, assunzione di farmaci o, più raramente, da
vaccinazioni. La diagnosi è clinica. Non esiste una terapia
specifica, trattandosi di una malattia benigna auto-limitantesi a
risoluzione spontanea. Alcuni autori hanno dimostrato che l'utilizzo
di corticosteroidi e/o di antistaminici non modifica la storia
naturale della patologia.
Figura
Glomerulonefrite
e neurite secondarie a sindrome vasculitica da overlap tra porpora di
Schönlein-Henoch e sindrome di Kawasaki: caso clinico
1Scuola
di Specializzazione in Pediatria, Università di Napoli
Federico II
2Dipartimento
di Nefrourologia, S.C. di Nefrologia e Dialisi, A.O.
Santobono-Pausilipon, Napoli
Caso
clinico. A. è un ragazzo di 14 anni che viene trasferito
presso la nostra struttura per macroematuria e proteinuria da altro
presidio ospedaliero dove era stato ricoverato circa un mese prima
per porpora e artralgie. Durante il precedente ricovero A. era stato
sottoposto ad appendicectomia per dolori addominali violenti. Alla
nostra osservazione il ragazzo si presenta in condizioni generali
scadenti, facies sofferente, marcata ipotonia muscolare agli arti
inferiori e superiori con grande difficoltà alla deambulazione
e alla prensione, petecchie agli arti inferiori, ipertensione
arteriosa, addome dolente con incisione chirurgica parzialmente
deiscente. Gli esami di laboratorio mostrano funzione renale nella
norma, marcata leucocitosi neutrofila, piastrinosi, microematuria
intensa e proteinuria di 3 g/die. Gli esami autoanticorpali, le
immunoglobuline sieriche, complemento, pannello virale (epatite,
TORCH, parvovirus B19), Vidal Wright risultano negativi o nella
norma. Il paziente, già in trattamento con prednisone (60
mg/die), lamenta forti dolori addominali, alla palpazione dell'addome
Murphy positivo: l'eco addome mostra colecisti infiammata e sludge
biliare, anse intestinali con parete ispessite. Il paziente inizia
terapia con acido ursodesossicolico. Per l'ipertensione arteriosa
vengono somministrati ACE-I e Ca antagonisti, con buon controllo dei
valori. Viene effettuata biopsia renale che mostra un quadro
immunoistologico di IgA nefropatia secondaria a porpora di S.H., II
stadio sec. Emancipator. Durante la degenza si osserva la comparsa di
desquamazione intensa delle mani e si assiste al peggioramento della
limitazione funzionale delle mani e degli arti superiori che sono
abbandonati ai lati del tronco. Vengono effettuati i potenziali
evocati motori e somatosensoriali e elettroneuromiografia che
mostrano alterazioni assonali a carico dei tronchi nervosi motori. La
RMN encefalospinale mostra un sistema ventricolare più ampio e
sottile iperintensità periventricolare e della sostanza
bianca. L'ecocardiografia non rileva alcuna alterazione delle
coronarie.
La
persistenza della invalidante sintomatologia neuritica ci ha indotto
a intraprendere terapia con ciclofosfamide orale che ha determinato
una lenta ma sicura ripresa funzionale della mani e della
deambulazione. Contemporaneamente anche i segni di nefrite sono
drammaticamente migliorati con la attenuazione della microematuria e
la quasi scomparsa della proteinuria (< 100 mg/die).
Discussione.
Il complesso quadro clinico esibito dal ragazzo è sicuramente
dominato dalla sindrome vasculitica con componenti tipiche
della porpora di Schönlein-Henoch (porpora, artrite, dolori
addominali e la nefrite immunoistologicamente caratterizzata) e altre
caratteristiche della sindrome di Kawasaki (la piastrinosi,
l'interessamento colecistico, congiuntivite rilevata all'intervista
anamnestica, desquamazione tardiva delle mani); infine l'elemento
atipico nel quadro delle sindromi citate è la neurite che è,
però, complicanza descritta di vasculiti sistemiche e non.
Università
di Padova
Un
bambino di sei anni, precedentemente sano, è stato valutato
presso il nostro Pronto Soccorso per comparsa di artralgie il giorno
successivo all'inizio di terapia antibiotica con amoxicillina per
un episodio febbrile della durata di 3 giorni. Le artralgie, esordite
inizialmente al gomito sinistro e agli arti inferiori, erano
associate a rash purpurico agli arti inferiori. È stata
pertanto posta diagnosi di porpora di Schönlein-Henoch. Il
paziente è stato dimesso in terapia antinfiammatoria con
ibuprofene (10 mg/kg ogni 8 ore).
Durante
il follow-up clinico l'artralgia si è diffusa, dall'iniziale
localizzazione al gomito sinistro, a ginocchia e anche
bilateralmente, associata a versamento articolare e comparsa di
dolore addominale. Visto il peggioramento dei sintomi addominali e
l'evoluzione di parte delle lesioni cutanee da purpuriche a
francamente bollose, dopo 10 giorni dall'esordio dei sintomi il
paziente è stato ricoverato.
Le
condizioni generali del paziente erano discrete, senza significativi
reperti all'esame obiettivo generale. Le lesioni cutanee erano
caratterizzate dalle classiche chiazze purpuriche palpabili, alcune
delle quali (localizzate agli arti inferiori) presentavano una bolla
centrale. Gli esami ematochimici (emocromo, funzionalità
renale ed epatica, elettroliti e profilo coagulativo) erano nella
norma, a eccezione del D-dimero (> 10 µg/ml FEU); anche la
PCR era lievemente aumentata (1,5 mg/dl) mentre il sangue occulto
fecale è risultato positivo in 3 differenti campioni.
Sierologie virali per EBV e parvovirus e batteriche per Mycoplasma
non hanno dimostrato infezioni attive. La coltura di una delle
lesioni, la coprocoltura e la ricerca di virus fecali sono risultate
negative.
Visto il
coinvolgimento addominale e la presenza di lesioni bollose, è
stata avviata terapia steroidea con prednisone (1 mg/kg/die),
associato a terapia antibiotica orale con amoxicillina-clavulanato
per 8 giorni totali.
Durante
la degenza il paziente si è mantenuto stabilmente apiretico;
il dolore addominale si è progressivamente risolto. Le lesioni
purpuriche sono gradualmente scomparse, con evoluzione delle bolle a
croste. Il paziente è stato dimesso in quarta giornata di
ricovero. La terapia steroidea è stata gradualmente scalata a
partire dal 7° giorno; le croste sono scomparse dopo circa 20
giorni senza lasciare cicatrici.
Discussione.
La porpora di Schönlein-Henoch (PSH) è la più
comune vasculite dell'infanzia.
È
una patologia multisistemica che interessa prevalentemente la cute,
le articolazioni, il tratto G-I e il rene, anche se altri organi
possono essere colpiti (SNC, testicoli, cuore, interstizio
polmonare).
La
diagnosi è nella maggior parte dei casi clinica, basata sulla
presenza dei segni e sintomi più frequenti all'esordio:
dolore addominale diffuso, associato o meno a emorragie intestinali
(più spesso con positività del sangue occulto fecale),
artriti o artralgie, coinvolgimento renale (ematuria e/o proteinuria)
e soprattutto porpora palpabile, più frequentemente
distribuita agli arti inferiori e alla regione glutea.
La
biopsia cutanea, che evidenza infiltrato granulocitario associato a
depositi di IgA all'immunofluorescenza, non è mandatoria per
porre diagnosi nei pazienti con manifestazioni cliniche tipiche,
mentre può essere necessaria nei pazienti con lesioni cutanee
atipiche e variabilità nei tempi di evoluzione della porpora.
La
terapia della PSH è prevalentemente di supporto (riposo,
idratazione orale), a cui può essere associato il trattamento
con FANS (ibuprofene, flurbiprofene, naprossene) in caso di
artralgie, e la terapia steroidea (prednisone 1 mg/kg/die) nelle
forme complicate (dolore addominale moderato-grave, coinvolgimento
renale con ematuria persistente).
A
differenza della forma adulta della malattia (peraltro rara), in cui
le lesioni purpuriche cutanee hanno un aspetto bolloso nel 16-60% dei
casi, la PSH bollosa è stata raramente descritta nei pazienti
pediatrici (< 2% dei casi). Questa presentazione insolita della
malattia può pertanto essere misconosciuta e causare errori al
momento della diagnosi. La presenza delle lesioni bollose non sembra
comunque avere un valore prognostico, essendo la PSH bollosa una
malattia benigna nella maggior parte dei casi, con risoluzione
completa a medio termine, proprio come la forma classica di PSH.
Nei pochi
casi descritti in letteratura di PSH bollosa, la terapia steroidea
sembra avere un ruolo nel ridurre la severità e l'estensione
delle lesioni bollose, come anche le sequele e le complicanze cutanee
(dolore, necrosi, sovrainfezione).
Conoscere
e saper riconoscere questa forma rara di PSH è pertanto utile
perché essa può rappresentare un'indicazione
all'inizio precoce del trattamento steroideo anche in assenza delle
forme ben note di coinvolgimento renale e intestinale.
1Scuola
di Specializzazione in Pediatria, Università di Napoli
Federico II
2Dipartimento
di Nefrourologia, S.C. di Nefrologia e Dialisi, A.O.
Santobono-Pausilipon, Napoli
Introduzione.
La porpora trobocitopenica idiopatica (PTI) in età pediatrica
ha un'incidenza che va da 1,9 a 6,4/100.000 bambini/anno1;
ci sono casi in cui però una porpora trombocitopenia
apparentemente idiopatica può essere manifestazione clinica di
una patologia più complessa.
Case
report. R. giunge alla nostra osservazione per riscontro
all'esame urine di microematuria e proteinuria. Nell'aprile 2009
in seguito alla comparsa di ecchimosi agli arti inferiori viene
riscontrata la presenza di piastrinopenia (25.000/?l). Condotta
presso la divisione di Onco-Ematologia del nostro PO, viene posta
diagnosi di PTI ed effettuata terapia corticosteroidea per circa un
anno con incremento dei valori piastrinici. Nel dicembre 2010
compaiono lesioni purpuriche agli arti inferiori e alle natiche con
artralgia ed edema a carico di mani e piedi, in tale occasione la
piccola effettua esame urine che evidenzia presenza di ematuria e
proteinuria. Eseguito un secondo esame urine che conferma il dato
patologico, si ricovera presso la Struttura Complessa di Nefrologia e
Dialisi dell'Ospedale Santobono-Pausillipon. R. all'ingresso ha
11 anni, pesa 54.500 (90° ct), è alta 157 cm (75-90°
ct), e ha una PA nella norma, sono buone le sue condizioni cliniche
generali. Da un approfondimento anamnestico si evince presenza di
rash malare in seguito a fotoesposizione. La storia di
trombocitopenia, artrite, rash malare e l'attuale coinvolgimento
renale orientano verso la diagnosi di lupus eritematoso sistemico,
confermata dalla positività degli anticorpi anti-nucleo e
degli anticorpi anti-DNA nativo. Durante la degenza le indagini
praticate sulle urine evidenziano una proteinuria importante (1700
mg/24 h) con ematuria a fondo scala; l'esame del sedimento urinario
mostra caratteristiche di nefrite attiva (emazie di tipo glomerulare,
cilindri di emazie, leucociti), mentre gli esami ematochimici mettono
in luce l'ipocomplementemia (riduzione del C3 e C4) e funzionalità
renale nella norma (GFR secondo Schwartz: 97 ml/min/1,73 m2/s).
Per eseguire la stadiazione istologica della nefrite R. viene
sottoposta ad agobiopsia renale eco-assistita che definisce una
nefrite lupica al III stadio. Pertanto inizia tempestivamente la
terapia con micofenolato mofetile e cortisone. a un mese dalla
dimissione si assiste alla negativizzazione della proteinuria, alla
riduzione dell'ematuria, alla normalizzazione della conta
piastrinica e della complementemia.
Conclusioni.
La trombocitopenia è una manifestazione clinica comune nel LES
(7-30%)2 e si presenta all'esordio della patologia nel
5% dei casi. Hazzan et al.3 sostengono che la maggior
parte dei bambini con PTI e ANA positivi non sviluppa LES e che la
produzione di ANA in corso di PTI può essere dovuta a
induzione da parte di patologie virali. Solo il 3,6% di bambini con
PTI sviluppa LES a distanza di 4,2 anni (media del follow-up),
soprattutto in soggetti di sesso femminile, età media di 12,7
anni e alto titolo di ANA. La piccola R. ha ricevuto diagnosi di LES
a distanza di due anni dall'esordio della piastrinopenia, non
conosciamo il titolo di ANA a quel tempo ma una loro positività
c'avrebbe indotto a ricercare nei mesi successivi altre
manifestazioni cliniche di LES con la possibilità di porre
diagnosi più precocemente.
Bibliografia
- Terrel DR, et al. The incidence of immune thrombocytopenic purpura in children and adults: a critical review of published reports. Am J Hematol 2010;85:174-80.
- Ziakas, et al. Lupus thrombocytopenia: clinical implications and prognostic significance. Ann Rheum Dis 2005;64:1366-9.
- Hazzan, et al. Risk factors for future development of Systemic Lupus Erythematosus in children with Idiopathic Thrombocytopenic Purpura. Pediatr Blood Cancer 2006;47:657-9.
Clinica
pediatrica, IRCCS pediatrico Burlo Garofolo, Trieste
N. è
una ragazza di 15 anni di origine indiana che da 6 mesi presenta
dolore addominale, diarrea alternata a stipsi e ulcere orali. Un mese
prima ha avuto un episodio di dolore e arrossamento oculare
all'occhio sinistro che è stato diagnosticato come uveite
anteriore acuta e trattato con terapia topica steroidea. Un anno
prima si era verificata la comparsa di lesioni nodulari dolorose agli
arti inferiori. La diagnosi era stata quella di eritema nodoso di
origine tubercolare in seguito alla forte reazione cutanea (ampia
zona di vescicolazione e necrosi cutanea) alla Mantoux che era stata
eseguita per indagare l'eziologia dell'eritema nodoso. La
radiografia del torace era negativa e non presentava altri segni
obiettivi di tubercolosi. È stata trattata con 6 mesi di
terapia antitubercolare, con risoluzione delle lesioni. Al momento
del ricovero nel nostro reparto presentava solo una modesta
dolorabilità alla palpazione profonda in fossa iliaca sinistra
e piccole ulcere orali. Le prove di laboratorio mostravano una VES di
78 mm/h e una calprotectina fecale di 74 mg/kg (v.n. < 15). Per
escludere la tubercolosi intestinale o una malattia infiammatoria
cronica sono state eseguite una colonscopia e un'endoscopia con
videocapsula che hanno dimostrato la presenza di afte ileali sparse.
La ricerca di M. tuberculosis è risultata negativa così
come la radiografia del torace. Nel complesso la diagnosi di
tubercolosi appariva poco probabile mentre risultavano soddisfatti i
criteri per la diagnosi di m. di Behçet (ulcere orali
ricorrenti, uveite, eritema nodoso). Il coinvolgimento intestinale
sotto forma di ulcere ileali è anche descritto nella m. di
Behçet. La ragazza è stata curata con cortisone per os
per 2 mesi con ottimo recupero. A posteriori, la forte reazione
cutanea alla Mantoux può essere spiegata come una reazione di
tipo patergico, tipica della m. di Behçet. Può essere
dunque opportuno ricordare la possibilità di una reazione
falsamente positiva alla Mantoux nei pazienti che presentano sintomi
compatibili sia con la TBC che con la m. di Behçet.
Clinica
pediatrica, IRCCS pediatrico Burlo Garofolo, Università
di Trieste
G. è
una bambina di 10 anni che si presenta per una storia di progressiva
astenia, inappetenza e calo ponderale associati a rilievo di anemia.
La sua storia clinica inizia due anni prima quando, a seguito di un
morso di zecca, compare una tumefazione linfonodale retrocervicale.
In quell'occasione era stata riscontrata una VES di 45 mm/h con
ematuria all'esame urine. Gli esami ripetuti a 3 mesi di distanza
avevano confermato il rialzo della VES (35) con lieve anemia (Hb 10,3
g/dl). Nei mesi successivi la bambina era stata bene e il dato non
era stato ulteriormente indagato. Nei 4 mesi precedenti il ricovero
nel nostro reparto, ha iniziato a presentare progressiva astenia e
inappetenza con calo ponderale (5 kg). All'ingresso si presentava
in discrete condizioni generali, niente da segnalare all'esame
obiettivo eccetto il pallore e la pressione arteriosa elevata (127/95
mmHg, > 95° percentile). Gli esami ematochimici rilevavano
anemia severa (Hb 6,7 g/dl) con reticolociti relativamente bassi (1%)
e importante elevazione della VES (96 mm/h). Normali la PCR,
l'aptoglobina e lo striscio periferico. Creatinina lievemente
aumentata (1,1 mg/dl). L'esame urine evidenziava proteinuria 4+ ed
ematuria glomerulare (220 GR/mm3). Sulla raccolta urine delle 24 h la
proteinuria risultava 2500 mg, non selettiva (indice di Cameron 1,5).
C3 e C4 nella norma, negativa la ricerca di ANA e anti-DNA. Positiva
invece la ricerca degli anticorpi p-ANCA (anticorpi
anti-mieloperossidasi) (47 U/ml). Negativi gli altri accertamenti
(aspirato midollare, radiografia del torace, ecografia addominale,
consulenza oculistica e cardiologica). Nel sospetto di una
glomerulonefrite da vasculite ANCA-associata è stata eseguita
una biopsia renale che ha evidenziato una glomerulonefrite
proliferativa extracapillare focalmente necrotizzante con depositi di
IgM all'immunofluorescenza. La diagnosi finale pertanto è
stata di micropoliangioite (MPA) con glomerulonefrite. È stata
avviata terapia con boli di cortisone a giorni alterni, seguita da
terapia con prednisone 50 mg/die e ciclo di ciclofosfamide per os
(100 mg/die per 3 mesi). La malattia a 3 mesi dalla diagnosi ha
risposto solo parzialmente alla terapia, con lieve riduzione della
proteinuria e dell'anemia.
Specializzanda
presso la Clinica Pediatrica dell'IRCCS pediatrico Burlo
Garofolo, Trieste
Vuoi citare questo contributo?