Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Febbraio 2010 - Volume XIII - numero 2
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Dolori
addominali ricorrenti da invaginazione intermittente.
Un
caso di sindrome di Peutz-Jeghers in una bambina di 13 anni
Dipartimento
di Pediatria, Università di Firenze, Ospedale Meyer, Firenze
Indirizzo
per corrispondenza: paolo.lionetti@unifi.it
RECURRENT
ABDOMINAL PAIN DUE TO INTERMITTENT INTUSSUSCEPTION.
A
CASE OF PEUTZ-JEGHERS SYNDROME IN A 13-YEAR-OLD GIRL
Key
words recurrent abdominal pain, Peutz-Jeghers Syndrome, small-bowel intussusceptions, intestinal polyposis Summary
Peutz-Jeghers
syndrome (PJS) is a rare, though well-described, hereditary
disorder characterized by mucocutaneous pigmentation and
hamartomatous polyps that tipically presents in the second decade
of life. The authors present a case of a 13 year-old girl who
presented with a 4-month story of
abdominal pain, located in the periumbilical region, initially
without associated symptoms and then followed by nausea and
vomiting. Utrasound scan and, then,
exploratory laparotomy revealed small-bowel intussusceptions due
to the presence of two jejunal polyps: resections and
anastomosis of the involved segment was done. Histopathology
revealed the presence of hamartomas and hyperpigmintation on her
lips were discovered: the young patient was diagnosed with
Peutz-Jeghers Syndrome. The diagnosis was confirmed by the
presence of a mutation of STK11 (LKB1)
gene. |
|
Descriviamo
il caso di una bambina che viene portata dal pediatra alletà
di 13 anni per una storia di dolori addominali periombelicali
ricorrenti iniziata tre mesi prima. Dallanamnesi familiare
emerge una storia di poliposi intestinale (nonno paterno) e di
carcinoma colico (nonno materno). Gli episodi di addominalgia
riferiti dalla bambina sono spesso mattutini, a risoluzione
spontanea e non associati ad altra sintomatologia. Le condizioni
cliniche della bambina sono buone, la crescita nella norma, lesame
obiettivo negativo. Il pediatra prescrive degli esami ematochimici di
routine, compresa la sierologia per celiachia, risultati negativi e
conclude per una natura funzionale del problema. Nel mese successivo,
tuttavia, gli episodi diventano più frequenti e, anche se mai
notturni, questa volta si accompagnano ad un segnale di allarme:
sono spesso seguiti da vomito.
Al di
fuori di tali episodi, la bambina appare in completo benessere. La
famiglia è molto tranquilla. A questo punto il pediatra
prescrive unecografia delladdome, che mette in evidenza
a livello epigastrico, la presenza di unansa intestinale a
pareti marcatamente ispessite al cui interno sembra apprezzabile
unimmagine di doppio lume. Il pediatra invia la bambina presso
il nostro Ospedale, dove viene eseguito un secondo controllo
ecografico che mette in evidenza la presenza di unimmagine a
coccarda, circondata da modesta falda fluida, che si risolve e
si riforma durante lindagine. Le anse coinvolte,
verosimilmente dellintestino tenue, appaiono modicamente
ispessite (3.5-4 mm) senza tuttavia significativa alterazione
strutturale e vascolare. Il reperto è indicativo di
uninvaginazione intestinale intermittente. Lesame
obiettivo della bambina è negativo e non vi sono
manifestazioni emorragiche. Considerata letà della
paziente sospettiamo uninvaginazione su diverticolo di Meckel
o su polipo, ma temiamo anche un linfoma. Durante la degenza, per
escludere la presenza di patologie di natura infiammatoria o
neoplastica, vengono effettuate ulteriori indagini di laboratorio.
Gli
indici di flogosi e gli esami ematochimci di routine risultano nella
norma, viene inoltre effettuato il dosaggio degli ASCA IgG e IgA
(esame che si riscontra positivo nel 60% dei casi di Malattia di
Crohn) e dei pANCA (anticorpi positivi nel 70% dei casi di colite
ulcerosa), entrambi risultati negativi; la calprotectina fecale
(marker di flogosi intestinale) è nella norma. Risultano
negativi il Ca 125 ed il Ca 19-9 Gica (importanti marker tumorali
comunemente utilizzati per la valutazione diagnostica e prognostica
di alcuni tipi di cancro, tra cui quelli del tratto gastroenterico),
nella norma lo striscio di sangue periferico, negativa anche la
ricerca del sangue occulto nelle feci in due occasioni. Vengono poi
eseguite indagini strumentali, tra cui una RM addominale ed una
ecografia con clisma con acqua, che confermano il quadro di
invaginazione intestinale intermittente dellintestino tenue.
Dal punto di vista clinico la ragazza risulta pressoché
asintomatica ad eccezione di episodi di vomito mattutino. Si comunica
ai genitori della bambina la necessità di una laparoscopia
esplorativa. I genitori vogliono prendere tempo: sono molto
perplessi, soprattutto in considerazione dellapparente
benessere della bambina. Alla fine decidono di ricondurre la figlia a
domicilio contro il parere dei sanitari. A distanza di una settimana
la bambina viene condotta durgenza in Pronto Soccorso con un
chiaro quadro sub-occlusivo caratterizzato da intenso dolore
addominale e vomito. Viene sottoposta ad intervento chirurgico di
laparotomia esplorativa, durante il quale viene repertata una doppia
invaginazione a carico dellintestino tenue su doppio polipo
per cui viene eseguita la resezione del tratto interessato (circa 50
cm) con confezionamento di anastomosi termino-terminale. Lesame
istologico del materiale prelevato documenta la natura amartomatosa
dei polipi asportati. Il decorso post-operatorio è regolare.
La bambina viene dimessa. Solo nei giorni successivi, in occasione di
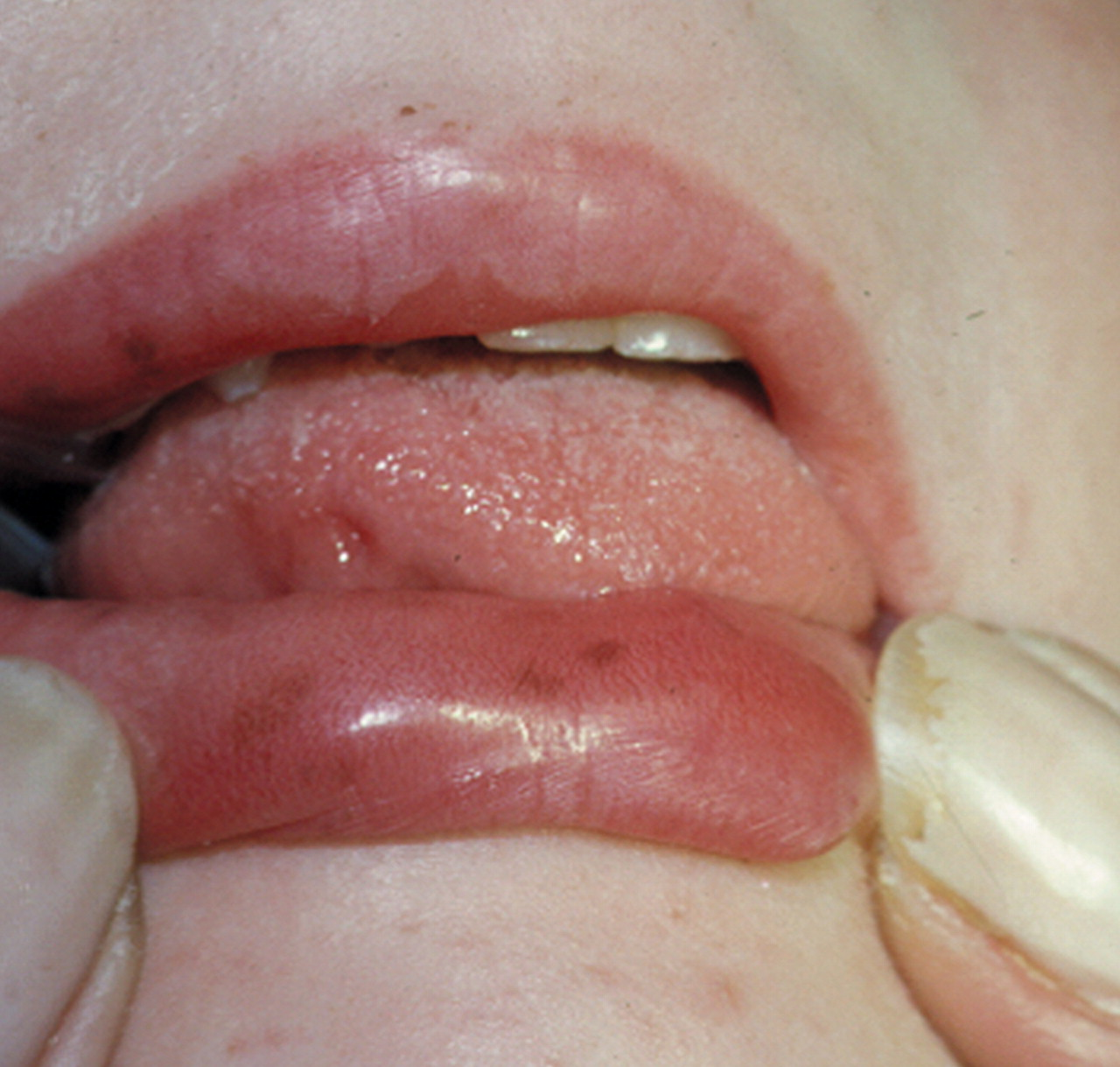
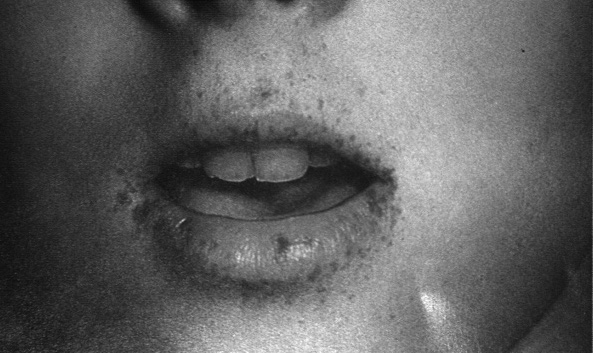
una visita ambulatoriale eseguita dopo la dimissione, vengono notate
alcune macchie iperpigmentate a livello della superficie interna
della mucosa labiale e qualche piccola lentiggine periorale, la cui
comparsa viene riferita graduale dopo i 5 anni di età.
Nel
sospetto di una sindrome di Peutz-Jeghers, viene eseguita una
valutazione genetica che conferma la presenza di una mutazione a
livello del gene STK11 (LKB1) localizzato sul cromosoma 19p.
I dolori
addominali ricorrenti DAR1,2 rappresentano
per il pediatra di famiglia ed i Pronti Soccorso degli Ospedali
Pediatrici una problematica di riscontro molto frequente. Nei bambini
dai 4 ai 14 anni si manifestano in una percentuale che va dal 4 al
25%. Nel 90% dei casi si tratta di dolori di natura funzionale:
lassenza dei classici segnali di allarme, le cosiddette
bandierine rosse red flags (Tabella 1) e la negatività
degli esami minimi di routine consente di identificarli come tali.
Non va tuttavia dimenticato che nel 10% dei casi sono dovuti ad una
causa organica, che va sempre necessariamente sospettata quando
appunto siamo in presenza di qualche segnale di allarme. Nel nostro
caso, ci troviamo di fronte ad una bambina con i classici dolori in
sede periombelicale, che si risolvono spontaneamente, mai notturni e
con periodi di completo benessere tra un episodio doloroso e laltro.
Inizialmente sono assenti elementi di preoccupazione e tutto fa
pensare ai soliti disturbi funzionali, come se ne vedono tanti nei
nostri ambulatori. Ad un certo punto, però, compare un sintomo
che ci porta a rivalutare il caso: gli episodi di dolore diventano
più importanti e, soprattutto, si associano a vomito. Per
questo vengono effettuate indagini più approfondite che ci
hanno permesso di giungere alla diagnosi di invaginazione intestinale
secondaria a poliposi nellambito di una sindrome di
Peutz-Jeghers.
Linvaginazione
intestinale, in generale, rappresenta in età adolescenziale
una causa poco comune di dolore addominale. Si presenta più
frequentemente, infatti, nel bambino (92% dei casi) con un picco
massimo di incidenza tra i 2 mesi ed i 2 anni di vita. Leziologia
e la sede variano in rapporto alletà: al di sotto dei 2
anni è quasi sempre idiopatica e di tipo ileo-colico, mentre
nelle età successive è generalmente secondaria ad una
patologia intestinale organica (diverticolo di Meckel, lesione
polipoide, neoplasia, vasculite) ed è di tipo ileo-ileale o
ileo-colico.
La
Sindrome di Peutz-Jeghers (PJS), a sua volta, rappresenta una rara
causa di invaginazione intestinale. È una malattia autosomica
dominante a penetranza variabile, la cui incidenza è stimata
intorno a 1 caso ogni 30000 nati. Risulta associata a mutazioni del
gene STK11/LKB1 (19p13.3)3,4, che vengono riscontrate
mediante test genetico in più del 70% dei casi di PJS
familiare ed in una percentuale variabile tra 30 e 70% nei casi di
PJS sporadica . Numerosi dati suggeriscono, tuttavia, una
eterogeneità genica, il verosimile coinvolgimento di altri
loci e il fatto che nel 35% dei casi risulta determinata da
mutazioni de novo.
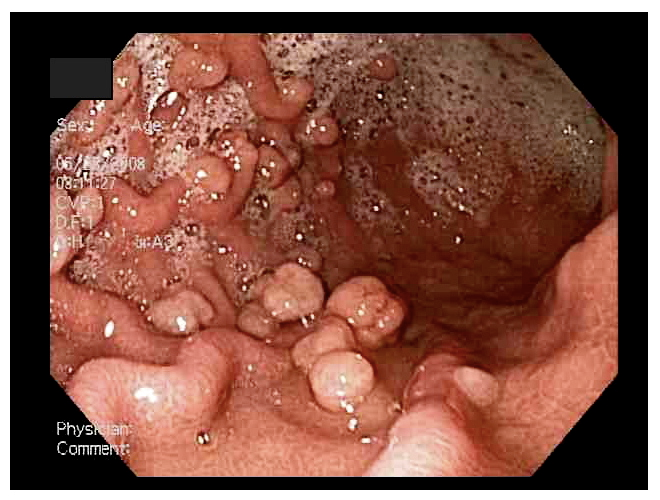
LA PJS è
caratterizzata dalla presenza di polipi, generalmente multipli, di
tipo amartomatoso a livello gastrointestinale associati a
pigmentazione melaninica muco-cutanea. Le lesioni iperpigmentate,
presenti nel 95% dei casi, si localizzano per lo più a livello
della mucosa labiale e della regione periorale, tendono a svilupparsi
entro i 5 anni di vita ed a scomparire gradualmente alla pubertà
o in età adulta. I polipi, di dimensioni variabili, sono
descritti in tutto il tratto gastroenterico (anche se sono più
frequentemente localizzati a livello dellintestino tenue, dove
si presentano fino al 96 % dei casi)5 e possono
determinare sanguinamento, dolore addominale, invaginazione, volvolo,
prolasso rettale recidivante. Nei soggetti senza storia familiare di
malattia, la diagnosi clinica può essere formulata sulla base
del riscontro di almeno due polipi intestinali amartomatosi (anche in
assenza delle pigmentazioni muco-cutanee), mentre nei pazienti con
familiarità accertata per PJS (parente di primo grado affetto)
è sufficiente la presenza delle lesioni iperpigmentate (anche
senza dimostrazione della presenza di polipi amartomatosi)5.
I pazienti affetti da PJS hanno, inoltre, un rischio aumentato di
sviluppare altre neoplasie (a carico del tratto gastrointestinale,
colecisti, pancreas, tiroide, seni nasali, polmoni, mammella ed
apparato genito-urinario)6,7: in questottica
risulta importante eseguire un follow-up accurato di prevenzione5-9.
Gli esami ematochimici (emocromo, markers tumorali) vanno eseguiti
annualmente dopo la diagnosi di SPJ, le indagini endoscopiche
intestinali (colonscopia+EGDS e videocapsula) possono essere
ripetute con una frequenza biannuale a partire dalletà
di 10 anni; lecografia testicolare e quella pelvica vanno
eseguite annualmente a partire rispettivamente dai 10 anni e dai 20
anni di età; la visita senologica deve essere ripetuta
annualmente dai 25 anni, lecografia mammaria (con eventuale
mammografia) può essere effettuata ogni 2-3 anni a partire dai
25 anni e lecografia addominale biannualmente a partire dai 30
anni. Gli intervalli di esecuzione delle varie indagini previste dal
follow-up possono naturalmente essere modificati sulla base della
sintomatologia clinica.
Segnali
di allarme Red Flags nei DAR |
|
Tabella
1. I segnali di allarme che devono far pensare a una patologia
organica quando un bambino si presenta con dolori addominali
ricorrenti (DAR)
- Baber KF, Anderson J, Puzanovova M, Walker LS. Rome II Versus Rome III Classification of Functional Gastrointestinal Disorders in Pediatric Chronic Abdominal Pain. Journal Pediatr Gastroenterol Nutr 2008;47:299-302.
- Zeiter DK, Hyams JS. Recurrent abdominal pain in children. Pediatr Clin North Am 2002;49:53-71.
- Buck JL, Harned RK, Lichtenstein JE, Sobin LH. Peutz-Jeghers sindrome. Radiographics 1992;12:365-387.
- Schreibman IR, Baker M, Amos C, McGarrity T. The hamartomatous polyposis syndromes: a clinical and molecolar review. Am J Gastroenterol 2005;100:476-90.
Sito Geneclinics. Amos CI, McGarrity TJ, Frazier ML. Peutz-Jeghers syndrome. Revisione aggiornata al 30 dicembre 2002.
Westerman AM, et al. Peutz-Jeghers sindrome: 78 years follow-up of the original family. Lancet 1999;353:1211-15.
- Giardiello FM, et al. Very high risk of cancer in familial Peutz-Jeghers sindrome Gastoenterology 2000;119:1447-53.
- McGarrity TJ, Kulin HE, Zaino RJ. Peutz-Jeghers syndrome. Am J Gastroenterol 2000;95:596-604.
- Centuori S, Martellossi S, Ammar L. Storia di una famiglia con sindrome di Peutz Jeghers: importanza dello screening strumentale anche nei soggetti asintomatici. Medico e Bambino pagine elettroniche 2003;6(9)
Centuori
S, Martelossi S, Ammar L. Storia di una famiglia con sindrome di
Peutz Jeghers: importanza dello screening strumentale anche nei
soggetti asintomatici. Medico
e Bambino
pagine elettroniche 2003;6(9)
Bua J,
Norbedo S, Martelossi S. Sangue e muco nelle feci. Medico
e Bambino pagine elettroniche 2006;9(2).
Vuoi citare questo contributo?