Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Febbraio 2009 - Volume XII - numero 2
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Una
plica cutanea sospetta... il primo passo verso la diagnosi di sindrome
di Klippel-Trenaunay
Pediatri
di famiglia, Napoli
Indirizzo
per corrispondenza: tom.montini@libero.it
A
suspicious cutaneous plica
the first step towards the
diagnosis of Klippel-Trenaunay syndrome
Key
words
Klippel-Trenaunay
syndrome, case report, diagnosis, guidelines
Summary
We
describe a case of Klippel-Trenaunay syndrome (KTS) diagnosed to
a one-year female child followed by our ambulatory of general
paediatrics. We diagnosed the KTS on the basis of a (pseudo)
hemihypertrophy of the legs and of a large capillary angioma of
the whole right leg. We also debate on the difficulty to find
clear guidelines for the diagnosis and follow-up of KTS in the
literature. |
La
piccola L. è nata da parto cesareo d'elezione a 39 settimane
di età gestazionale dopo una gravidanza normocondotta. Il peso
alla nascita è stato di 2630 g con indice di Apgar 8 (ad 1')
e 9 (a 5'). Alla prima visita effettuata presso il nostro
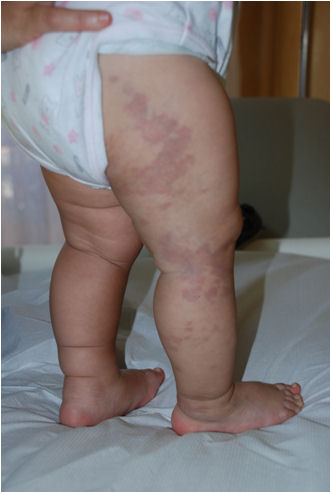
ambulatorio viene riscontrato un angioma capillare piano
superficiale, serpiginoso, di grosse dimensioni, esteso a tutto
l'arto destro (Figura 1), e un altro di
piccole dimensioni a livello della palpebra sinistra. Alle visite
successive la crescita della bambina è apparsa sempre
soddisfacente; allattamento materno, normale svezzamento, adeguato
sviluppo psicomotorio.
L. viene
al nostro ambulatorio all'età di un anno per un bilancio di
salute. La mamma ha notato una piega cutanea al terzo distale della
gamba sinistra, asimmetrica rispetto alla destra (Figura
1) e chiede se è normale. Alla visita la bambina è
in perfette condizioni generali e la piega sembra effettivamente
strana per la sua asimmetria rispetto all'arto
controlaterale. L'impressione è che i due arti siano
lievemente diversi di dimensione, e la piega sembra dovuta ad una
maggiore presenza di tessuto sottocutaneo; non si avverte pastosità
alla digitopressione, non c'è storia di edemi; i polsi
femorali sono normopulsanti da entrambi i lati. Decidiamo di misurare
gli arti: l'arto sinistro è leggermente più lungo del
destro (circa 4-5 mm), la circonferenza della coscia e della gamba
leggermente più grandi e anche il piedino è di pochi
millimetri più lungo del controlaterale.
Ci
prendiamo un giorno per studiare
. angiomatosi più
emiipertrofia è uguale a sindrome di Klippel-Trenaunay. La
consulenza genetica conferma la diagnosi.
La
sindrome di Klippel-Trenaunay (KTS), nota anche come sindrome
angio-osteoipertrofica, è descritta come una malattia
rara. In letteratura non siamo riusciti a trovare alcun accenno
all'incidenza, ma tra il 1970 e il 2005 presso la Mayo Clinic ne
sono stati osservati 218 casi1, e un altro lavoro ne
descrive oltre 700 casi2, quindi, tanto rara non
sembrerebbe. La triade di manifestazioni cliniche, così come
descritte originariamente da Maurice Klippel e Paul Trenaunay nel
1900, include malformazioni capillari cutanee, ipertrofia ossea e dei
tessuti molli e varici venose, anche se la triade coesiste soltanto
in circa il 60% dei casi3. Successivamente Parkes Weber
aggiunse alla descrizione la presenza di fistole arterovenose, e dopo
una lunga diatriba in letteratura, oggi la sindrome di Parkes-Weber è
considerata un'entità separata che include la triade della
KTS associata a fistole arterovenose.
La
malformazione vascolare è presente alla nascita e di
solito coinvolge un arto, preferenzialmente inferiore (come nel caso
della nostra piccola L.); più raramente coinvolge più
arti, il tronco, il volto o, raramente, il corpo intero3,4.
La malformazione vascolare (capillari telengectasici dilatati) è
tipicamente rappresentata da un nevo color vinoso, generalmente
localizzato nell'area ipertrofica (come nel nostro caso), che a
differenza degli emangiomi non ha la tendenza a regredire
spontaneamente con l'età5. Va sottolineato che,
mentre la maggior parte degli studi è concorde nel ritenere
che la malformazione vascolare sia sempre presente, uno studio su 614
pazienti ne riporta la presenza soltanto in un terzo dei casi2.
Le
varicosità venose diventano invece tipicamente evidenti
omolateralmente alla malformazione vascolare dopo che il bambino ha
iniziato a camminare. Anche su questo punto la letteratura è
discorde: varicosità venose compaiono dal 50 al 100% dei casi,
ma non è ben chiaro fino a che età possano iniziare a
comparire (nella nostra bambina, a tutt'oggi non sono evidenti).
Tali varicosità possono riguardare anche l'intero arto
inferiore (in alcuni pazienti è descritta la vena di
Klippel-Trenaunay) che compare alla nascita e si estende dal piede
fino alla regione gluteale, drenando nella femorale profonda o nel
sistema iliaco6. Nel nostro caso abbiamo eseguito una
valutazione vascolare ecodoppler degli arti inferiori che non ha
mostrato anomalie. Sono state descritte (l'incidenza non è
condivisa) varici venose negli organi interni (vescica, colon).
Un'ecografia addominale (difficile dire quanto sia in assoluto
dirimente) non ha mostrato anomalie nella piccola L.
Diversi
lavori riportano complicanze vascolari, da emorragie (anche
severe) a tromboflebiti, fino ad embolie polmonari. La sensazione è
che la severità di tali complicanze sia inversamente
proporzionale alla loro frequenza; in ogni caso, anche se non vi sono
specifiche indicazioni in letteratura su questo punto, abbiamo
ritenuto opportuno escludere, nella nostra piccola, la presenza di
predisposizione trombofilica che potrebbe render più frequenti
o severe tali complicanze, attraverso la valutazione della proteina C
ed S, dell'antitrombina III, del fattore V di Leyden e della
mutazione G20210A della protrombina (che non sono risultati
alterati), ben consapevoli di aver esplorato soltanto una piccola
parte dei fattori genetici di tipo protrombotico.
L'ipertrofia
ossea e/o dei tessuti molli è un altro elemento tipico
della KTS, e riconosce una patogenesi non nota ma indipendente
dall'alterazione vascolare. E' talora evidente alla nascita, ma
spesso si apprezza dopo il primo anno di vita (come nel caso della
nostra piccola L.); l'ipertrofia ossea è spesso causa di un
allungamento dell'arto (più raramente di un accorciamento).
Nel nostro caso una radiografia comparativa degli arti inferiori ha
mostrato un'uguale lunghezza dei segmenti ossei permettendo di
attribuire la dismetria alla sola ipertrofia dei tessuti molli. Nei
casi descritti in letteratura, l'emiipertrofia è tipicamente
omolaterale rispetto alla lesione angiomatosa. Il caso descritto è
in questo senso atipico poiché l'ipertrofia, per quanto
lieve, è localizzata all'arto controlaterale, ma anche
questa è un'evenienza possibile.
Altre
caratteristiche, meno costanti, della malattia sono3:
- macrocefalia con viso asimmetrico (assente nella nostra piccola);
- emangiomi del volto (idem);
- glaucoma (una visita oculistica nel nostro caso ha escluso glaucoma e cataratta); andrà ripetuta? Con che cadenza? Dalla letteratura non è chiaro se queste complicanze possono comparire tardivamente;
- mani e piedi grandi con macrodattilia (assenti nel nostro caso); osteosclerosi ed osteoperosi, calcificazioni endocraniche (escluse nel nostro caso da una radiografia del cranio);
- edema ed ulcere cutanee (assenti nella piccola L.);
- complicanze renali, inizialmente ritenute rare, poi documentate in circa il 30% dei pazienti; una review su una rivista del settore le descrive fin troppo in dettaglio; peccato che l'unica indicazione che non si riesce a evincere da quel lavoro (retrospettivo) è il tipo di monitoraggio che andrebbe effettuato ad un nuovo paziente, e soprattutto se vi è un'età in cui si potrà abbassare la guardia.
Molto
interessante la genetica della KTS: tra le tante teorie proposte, e
tra l'eterna diatriba tra i fautori della predisposizione genetica
(concordanza in gemelli monozigotici, casi familiari multipli, etc.,)
e i fautori della malattia sporadica (discordanza in gemelli
monozigotici e scarsa incidenza di casi familiari multipli!)7
abbiamo imparato che esiste un'eredità di tipo
paradominante8; in altri termini gli individui
affetti dalla sindrome nascerebbero con una mutazione in eterozigosi
a livello di un gene codificante una proteina coinvolta con
l'angiogenesi9 o con la vasculogenesi, ma la malattia si
manifesta soltanto nei casi in cui si verifica una seconda mutazione
somatica (ciò spiegherebbe sia il pattern a mosaico
riscontrato nella maggior parte dei pazienti, sia gli sporadici casi
familiari). Nel caso della nostra bambina, l'anamnesi familiare è
risultata negativa.
La nostra
difficoltà non è stata quella di formulare la diagnosi
(in questo le moderne risorse d'aggiornamento ci sono state
preziose) ma trovare delle linee guida univoche e chiare (in
letteratura, o presso qualche struttura di secondo o terzo livello)
che ci suggerissero il monitoraggio più adeguato delle
possibili complicanze della malattia. Ottenuta la diagnosi siamo
andati in cerca di qualche ausilio bibliografico che ci guidasse
nella gestione della piccola paziente. Una ricerca in Medline
non ha evidenziato nessun lavoro di metaanalisi sulla sindrome, e
delle circa 90 review pubblicate sulla malattia, nessuna di esse
descrive in maniera organica la gestione clinica della KTS e delle
sue eventuali complicanze, e nessuna review è presente su
riviste di pediatria generale. Medico e Bambino dedica un
piccolo paragrafo alla malattia nel corso di un aggiornamento
monografico intitolato Emangiomi e malformazioni vascolari di
oltre 10 anni fa10. La maggior parte dei lavori si
sofferma su aspetti specifici della sindrome, come quelli di
interesse dermatologico, di chirurgia plastica, sulle complicanze
urologiche o ortopediche, sulla diagnostica radiologica o sulle
peculiarità genetiche della malattia.
Il
management della KTS, così come le indagini diagnostiche
ausiliarie da effettuare dovrebbe essere personalizzato, e guidato
dalla valutazione clinica multidisciplinare del paziente3.
Il trattamento è generalmente conservativo.
In
conclusione: la piccola L. presenta (per ora) soltanto l'angiomatosi
e l'ipertrofia dell'arto inferiore sinistro; riteniamo che il
profilo delle indagini effettuate sia sufficiente ad escludere (per
ora) le più comuni complicanze della malattia.
Figura
1. Angioma capillare piano superficiale, serpiginoso, di grosse
dimensioni, esteso a tutto l'arto destro
Bibliografia
- Husmann DA, Rathburn SR, Driscoll DJ. Klippel-Trenaunay syndrome: incidence and treatment of genitourinary sequelae. J Urol 2007;177:1244-9.
- Servelle M. Klippel and Trenaunay's syndrome, 768 operated cases. Ann Surg 1985;201:365-73.
- Kihiczak GG, Meine JG, Schwartz RA, et al. Klippel-Lindau syndrome: a multisystem disorder possibly resulting from a pathogenic gene for vascular and tissue overgrowth. Int J Dermatol 2006;45:883-90.
- Berry SA, Peterson C, Mize W. Klippel-Trenaunay syndrome. Am J Med Genet 1998;79:319-26.
- Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg 1982;69:412-22.
- Roebuck DJ. Pictorial review: the imaging features of lower limb Klippel-Trenaunay syndrome. Clin Radiol 1994;49:346-60.
- Aelvoet GE, Jorens PG, Roelen LM. Genetic aspects of the Klippel-Trenaunay syndrome. Br J Dermatol 1992;126:603-7.
- Happle R. Klippel-Trenaunay syndrome: is it a paradominant trait? Br J Dermatol 1993;128:465-6.
- Tian XL, Kadaba R, You SA, et al. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature 2004:427:640-5.
- Dalmonte P, Bava G, Camassa N, Zannini L. Emangiomi e malformazioni vascolari. Medico e Bambino 1997;5:293-300.
Vuoi citare questo contributo?