Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Marzo 2008 - Volume XI - numero 3
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Eteroplasia
ossificante progressiva: una nuova mutazione del gene GNAS1 e review
della letteratura
Dipartimento
Integrato Materno-Infantile, Università di Modena e Reggio
Emilia
Indirizzo
per corrispondenza: sissimattia@katamail.com
Progressive
osseous heteroplasia: a new mutation in the gnas1 gne and review
of the literature
Key
words
Progressive
Osseous Heteroplasia, GNAS1, heterotopic ossification,
calcification
Summary
Progressive
osseous heteroplasia (PHO) is a recently described genetic
disorder of mesenchymal differentiation characterized by dermal
ossification during infancy and by progressive heterotopic
ossification of cutaneous, subcutaneous and deep connective
tissues during childhood. We report a case of progressive osseous
heteroplasia resulting from a de novo mutation in the GNAS1 gene
with only the mutations of the GNAS1 gene reported so far in
progressive osseous heteroplasia. This new mutation helps extend
the genotype-phenotype correlation further |
L'eteroplasia
ossea progressiva (PHO) è una malattia genetica,
caratterizzata da una differenziazione mesenchimale, con
ossificazioni dermiche durante l'infanzia e una progressione delle
ossificazioni eterotopiche a livello cutaneo, sottocutaneo e del
tessuto connettivo. Noi descriviamo un paziente con eteroplasia ossea
progressiva dovuta ad una nuova mutazione del gene GNAS1. Sono state
riportate, finora, solo 10 mutazioni del gene GNAS11.
Questa nuova mutazione può aiutare a comprendere meglio la
correlazione tra genotipo-fenotipo dell'eteroplasia ossea
progressiva.
CASO
CLINICO
Il nostro
paziente è un bambino di 18 mesi nato in Italia da genitori
albanesi.
E' il
primogenito di genitori non consanguinei, nato alla 34° w + 4
giorni, da parto cesareo per febbre materna, dopo gravidanza
normodecorsa. I parametri auxologici alla nascita evidenziavano un
grave ritardo di crescita: peso g 1805 (<< 3°p); lunghezza
cm 44 (<<3°p); c. cranica cm 29,8 (< 3°p). Non erano
presenti dismorfiismi cranio-facciali e l'assetto neurologico era
adeguato per l'età. L'anamnesi familiare era negativa per
malattie genetiche. Alla nascita sono state riscontrate lesioni
eritemato-papulose localizzate al dorso, in seguito diffuse al
resto del corpo con evoluzione di alcune papule in noduli ossificati
sottocutanei.
All'età
di 10 mesi, giungeva alla nostra attenzione, per il persistere del
quadro cutaneo.
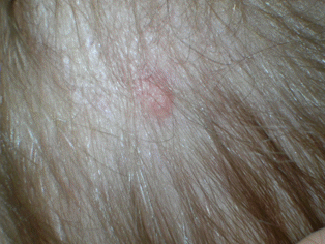
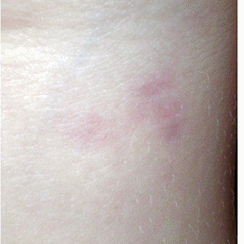
La cute
integra e normoelastica, mostrava diverse papule e noduli,
asintomatici, sparsi su tutto il corpo: tronco, arti, cuoio capelluto
(Figura1) con esclusione del volto. Tali
lesioni presentavano un colore variabile dal rosa al violaceo, ed
alcune erano palpabili. Le dimensioni variavano da 0,5 a 1,5 cm di
diametro (Figura 2). L'accrescimento
staturo-ponderale-cranico ha mantenuto il percentile della nascita,
con peso e circonferenza cranica inferiore al 3°p, altezza tra il
3° e 10°p, mentre lo sviluppo psico-motorio è
risultato sempre adeguato per l'età. Gli esami ematochimici
eseguiti per valutare la funzionalità tiroidea, renale,
epatica, i livelli sierici di Ca, P, PTH, vit D, la ricerca di
autoanticorpi e l'esame urine, sono risultati tutti nella norma.
La
biopsia cutanea della cute del dorso, rilevava a livello del
derma scarsissimo infiltrato linfocitario perivascolare,
calcificazioni e aspetti di ossificazione nel derma reticolare,
prevalentemente a disposizione periannessiale (Figura
3).
Tali
aspetti erano suggestivi di varie patologie: osteomatosi congenita in
placca, osteodistrofia ereditaria di Albright, eteroplasia ossea
progressiva e fibrodisplasia ossificata progressiva.
In
considerazione del normale sviluppo psicofisico, dei risultati
negativi degli esami e dell'esito della biopsia cutanea, veniva
ipotizzata la diagnosi di eteroplasia ossea progressiva (POH).
La
diagnosi è stata confermata dall'indagine genetica del gene
GNAS1 sul cromosoma 20 risultata positiva per una mutazione mai
descritta in letteratura, consistente in una delezione di una base
nell'esone 7, responsabile di un frameshift con codone di stop
prematuro pochi nucleotidi dopo.
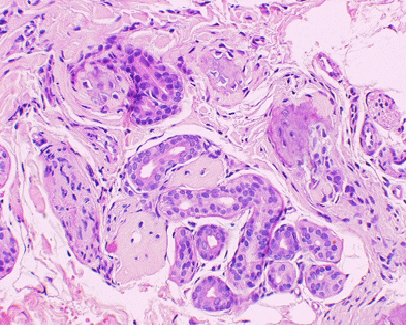
Figura
3. Biopsia della cute del dorso: aspetti di ossificazione nel
derma, attorno alle ghiandole sudoripare (ematossilina e eosina100x)
DISCUSSIONE
L'eteroplasia
ossea progressiva è una rara malattia autosomica
dominante, descritta per la prima volta da Kaplan nel 1994. Alcuni
casi di PHO sono sporadici, come il nostro paziente, mentre altri
sono familiari1.
Nel 1998
Urtizberea et al. riportano che la mutazione del gene GNAS1
responsabile della PHO è trasmessa con modalità
autosomica dominante. Nel 2002 in 13 casi su 18 di PHO (Tabella
1) è stata identificata una ereditarietà paterna2.
Nel nostro caso si tratta di una mutazione spontanea in quanto
l'indagine genetica effettuata sui genitori è risultata
negativa.
L'esatta
funzione del gene GNAS1 posto sul cromosoma 20 è ancora
sconosciuta, comunque, sembra codifichi la subunità alfa della
proteina G stimolatrice implicata nel controllo negativo della
osseogenesi.
Tale
patologia è caratterizzata da ossificazioni dermiche durante
l'infanzia e da una progressione delle ossificazioni eterotopiche a
livello cutaneo, sottocutaneo e del tessuto connettivo.
Generalmente
i primi sintomi compaiono durante l'infanzia e consistono in
piccole papule che precedono le papule e noduli ossificati
costituiti da osso eterotopico nel derma reticolare, col tempo
convergono a formare delle placche con coinvolgimento del tessuto
connettivo includendo eventualmente il tessuto muscolare, i tendini e
legamenti. La distribuzione dei noduli ossei è variabile
(Tabella 1), può essere diffusa come
nel nostro paziente o prediligere delle zone particolari, soprattutto
gli arti.
Gli esami
di laboratorio generalmente risultano normali; possono essere
transitoriamente elevati i livelli di fosfatasi alcalina, e i valori
della LDH e CPK in seguito alla deposizione di osso nella cute e
muscoli.
La
diagnosi si fonda sul riscontro dei seguenti elementi:
- Noduli duri sottocutanei
- Progressione dell'ossificazione verso i tessuti profondi
- Quadro istologico di ossificazione membranosa
- Esami emato-chimici ed endocrinologici normali
- Aspetto morfologico ed intelligenza normali
- Assenza di alterazioni scheletriche primitive
- Mutazione inattivante del gene GNAS, ma può anche mancare
- Familiarità che può mancare in presenza di mutazione spontanea
La
diagnosi differenziale della PHO deve essere posta con la
fibrodisplasia assificante progressiva (FOP), osseodistrofia
ereditaria di Albright (AHO) (Tabella 2) e
osteoma cutis in placche.
La
fibrodisplasia assificante progressiva nel nostro paziente è
stata ritenuta improbabile considerando l'assenza degli elementi
caratteristici della malattia quali: alluce valgo congenito,
microdattilia, noduli dolenti, pronunciata proliferazione di
fibroblasti coinvolgente le fibre muscolari, inoltre la FOP presenta
una ossificazione endocondrale piuttosto che intramembranosa come
nella PHO.
Anche
l'osteodistrofia ereditaria di Albright è stata
ritenuta improbabile perché ad eccezione della bassa statura,
risultavano assenti le frequenti anomalie a cui è associata
come obesità, ritardo mentale, brachidattilia e resistenza
all'azione del paratormone (pseudoipoparatiroidismo (PHP) o
pseudopseudoipoparatiroidismo). Sono state riportate occasionalmente
alcune ossificazioni eterotopiche nella AHO, e la documentazione di
due pazienti con AHO che mostravano ossificazioni eterotopiche
astese, suggeriva una comune base genetica con la PHO. L'AHO è
causata da un inattivazione eterozigote del gene GNAS1, ma a
differenza della PHO presenta una ereditarietà materna, che
causa anche PHP, mentre l'ereditarietà paterna causa il
fenotipo AHO senza resistenza ormonale
(pseudopseudo-ipoparatiroidismo). Quindi mutazioni che causano PHP
quando ereditate dalla madre possono causare PHO se ereditate dal
padre. Esiste comunque la possibilità che le due malattie PHO
e PHP possano coesistere3.
Le
malattie del connettivo (dermatomiosite, polimiosite, LES,
sclerosi sistemica), sebbene la presenza di calcificazioni e
ossificazioni raramente hanno esordio dalla nascita, sono state
escluse sulla base dei normali test di laboratorio; mentre l'osteoma
cutis in placche differisce solo nella natura progressiva della PHO.
La
severità e morbilità della PHO dipende dalla sede ed
estensione delle ossificazioni eterotopiche. La malattia non
interessa mai gli organi interni, quindi la vita del paziente non è
compromessa ma la qualità dipende dall'entità
dell'ossificazione e dalle zone interessate. Eventuali traumi non
sembrano precipitare o esacerbare le lesioni. Attualmente il nostro
paziente non presenta nessuna limitazione funzionale, e conduce una
vita paragonabile ai suoi coetanei.
Possibili
complicanze sono le anchilosi delle articolazioni coinvolte con
scarso accrescimento dell'arto, ulcerazione della cute, soprattutto
nelle zone di maggior pressione, con fuoriuscita di materiale osseo e
possibili infezioni estremamente dolorose. Aynaci et al.4
descrivono il caso di un paziente affetto da PHO con anchilosi di
entrambe le articolazioni degli arti superiori già all'età
di 5 anni.
Attualmente
non ci sono trattamenti validi o misure preventive. Il trattamento
è sintomatico, un ruolo importante è svolto dalla
fisioterapia per mantenere la mobilità delle articolazioni.
Dalla letteratura si apprende che due bambini subirono delle
amputazioni [uno di un arto inferiore5 ed un'altro di un
dito indice6] in seguito al ritardo di crescita severo e
perdita funzionale completa della parte affettata.
La
rimozione chirurgica dei noduli, determina delle recidive in molti
pazienti, soprattutto in presenza di ossificazioni eterotopiche
diffuse, piuttosto che localizzate7. Questo aspetto è
molto importante per il genetista, dermatologo, pediatra, e
ortopedico al fine di evitare trattamenti inefficaci e proporre un
counselling più efficace. La prognosi a lungo termine è
difficile da stabilire, poiché sono pochi i pazienti che sono
stati seguiti oltre l'adolescenza, comunque in questi pochi
pazienti la malattia ha presentato una progressione più lenta
durante l'età adulta.
In
conclusione questa nuova mutazione può aiutare a comprendere
meglio la correlazione tra genotipo-fenotipo dell'eteroplasia ossea
progressiva e a migliorare lo screening per una diagnosi prenatale.
Caso |
Referenza |
Età
alla diagnosi e sesso |
Area
di ossificazione |
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19 |
Edmonds
et al.8
Foster
et al.9
Foster
et al.9
Gardner
et al.10
Kaplan
et al.5
Kaplan
et al.5
Schmidt
et al.6
Athanasou
et al.11
Rodriguez-Jurado
et al.12
Rosenfeld,
Kaplan13
Rosenfeld,
Kaplan13
Miller
et al.14
Miller
et al.14
Urtizberea
et al.15
Stoll
et al.16
Chan
et al.1
Aynaci
et al.4
Gelfand
et al.3
Nostro
studio |
3,5
anni F
6
settimane F
Non
nota F
3
mesi F
18
mesi F
23
mesi F
9
anni F
6
settimane F
18
mesi F
6
anni M
3
anni M
6
mesi F
6
mesi F
1
mese F
2
anni F
9
mesi F
5
anni F
4
mesi M
14
mesi M |
Ascella
sinistra, braccio,
avambraccio,
mano e
dita.
Spalla
e gamba sinistra
Arto
sinistro
Fianco
destro, ginocchio e caviglia
Arto
inferiore sinistro
Arti
superiori
Regione
lombare destra paraspinale, spalla destra,regione pelvica destra.
Arto
inferiore sinistro, fianco e parete addominale
Tallone
sinistro, fossa
poplitea
e natica sinistra
Piede
e caviglia destra
Arto
inferiore sinistro
Arti
Arti
Tronco,
arti inferiori
Piedi,
ginocchia, caviglie, mani e sede sinistra della pelvi
Tronco
e arti
Arti
superiori
Coscia,
caviglia
Dorso,
arti, cuoio capelluto. |
Caratteristiche |
PHO |
FOP |
AHO |
Sesso
Trasmissione
genetica
Malformazioni
congenite
dell'alluce
Rash-papulare
congenito
Ossificazioni
cutanee
Ossificazioni
subcutanee
Ossificazioni
muscolari
Progressione
delle ossificazioni nel connettivo
Severa
limitazione alla mobilità
Ossificazioni
ectopiche dopo
i.m.
Ossificazione
Terapie
risolutive
|
Femmina=maschi
Autosomica
dominante
_
+
+
+
+
+
+
_
intramembranosa
_ |
Femmina=maschi
Autosomica
dominante
+
_
_
_
+
_
+
+
indocondrale
_ |
Femmina=maschi
Autosomica
dominante
_
_
±
±
_
_
_
_
intramembranosa
_ |
BIBLIOGRAFIA
1. Chan
I, Hamanda T, Hardman J, McGrath A, Child FJ. Progressive osseous
heteroplasia resulting from a new mutation in the GNAS1 gene. Clin
Exp Dermatol 2004;29(1):77-80.
2. Shore
EM, Ahn J, De Beur SJ, et al. Paternally inherited inactivating
mutations of the GNAS1 gene in progressive osseous heteroplasia. N
Engl J Med.2002;346(2):99-106.
3.
Gelfand IM, Hub RS, Shore EM, Kaplan FS, Dimeglio LA. Progressive
osseous heteroplasia-like heterotopic ossification in a male infant
with pseudohypoparathyroidism type Ia: a case report. Bone
2007;40:1425-28.
4.
Aynaci, Osman MD, Aynaci M, et al. Progressive osseous heteroplasia.
A case report and review of the literature. J Pediatr Orthop B
2002;11(4):339-42.
5. Kaplan
FS, Craver R, MacEwen GD, et al. Progressive osseous heteroplasia: a
distinct developmmental disorder of heterotopic ossification:Two new
case reports and follow-up of three previously reported cases. J Bone
Joint Surg 1994;74A:425-36.
6.
Schmidt AH, Vincent KA, Aiona MD. Hemimelic progressive osseous
heteroplasia. A case report. J Bone Joint Surg Am 1994;76:907-12.
7. Kaplan
FS, Shore EM. Progressive osseous heteroplasia. J Bone Mineral Res;
2000; 15: 2084-94.
8.
Edmonds HW, Coe HE, Tabrah FL. Bone formation in skin and muscle:
localized tissue malformation or heterotopia. J Pediatr
1948;33:618-23.
9. Foster
CM, Levin S, Levine M, et al. Limited dermal ossification: Clinical
features and natural history. J Pediatr 1986;109:71-6.
10.
Gardner RJM, Yun K, Craw SM. Familial ectopic ossification. J Med
Genet 1988;25:113-7.
11.
Athanasou NA, Benson MKDA, Brenton DP, Smith R. Progressive osseous
heteroplasia: a case report. Bone 1994;15:471-75.
12.
Rodriguez-Jurado R, Gonzalez-Crussi F, Poznanski AK. Progressive
osseous heteroplasia, uncommon cause of soft tissue ossification: A
case report and review of the literature. Pediatr Pathol Lab Med
1995;15:813-27.
13.
Rosenfeld SR, Kaplan FS. Progressive osseous heteroplasia in male
patients. Two new case reports. Clin Orthop 1995;317:24345.
14.
Miller ES, Esterly NB, Fairley JA. Progressive osseous heteroplasia.
Arch. Dermatol 1996;132:787-91.
15.
Urtizberea JA, Testart H, Cartault F, et al. Progressive osseous
heteroplasia. J Bone Joint Surg Br 1998; 80B:768-71.
16. Stoll
C, Javier MR, Bellocq JP. Progressive osseous heteroplasia: an
uncommon cause of ossification of soft tissues. Annales de genetique
2000;43:75-80.
Vuoi citare questo contributo?