Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Ottobre 2011 - Volume XIV - numero 8
M&B Pagine Elettroniche
Caso contributivo
Una
speciale anemia sideropenica
1Ematologia,
Dipartimento di scienze pediatriche, Università di
Torino
2Clinica Pediatrica, dipartimento materno-infantile, Università degli studi di Novara
3Fondazione IRCCS Ca Granda Policinico, Università di Milano
2Clinica Pediatrica, dipartimento materno-infantile, Università degli studi di Novara
3Fondazione IRCCS Ca Granda Policinico, Università di Milano
Indirizzo
per corrispondenza: annamartina77@yahoo.it
A case of iron-refractory iron deficiency anemia
Key
words Microcytic
anemia, Abnormal iron absorption and iron utilization, Autosomal
recessive inheritance
Abstract
Iron-refractory
iron deficiency anemia (IRIDA) is an autosomal recessive disorder
characterized by: congenital hypochromic, microcytic anemia, very
low mean corpuscular erythrocyte volume, low transferring
saturation, poor response to oral iron supplementation (abnormal
iron absorption) and partial response to parenteral iron therapy
(abnormal iron utilization).
IRIDA
has recently been shown to be caused by mutations in the gene
TMPRSS6 (located on the chromosome 22), which encodes a
transmembrane serine-protease expressed by the liver, which
modulates hepcidin. Hepcidin promotes the internalization and
degradation of ferroportin in lysosomes. As a consequence, both
iron absorption from the intestine and iron release from
macrophage stores are inhibited. |
L'anemia
ferro-carente ferro-refrattaria (IRIDA) è un'anemia
ipocromica microcitica congenita, a trasmissione autosomica recessiva
con MCV molto basso, bassa saturazione della transferrina, scarsa
risposta al ferro somministrato sia per via orale sia per via
parenterale.
L'epcidina
è un peptide, prodotto dal fegato, che controlla il
metabolismo del ferro promuovendo l'internalizzazione e la
degradazione della ferroportina nei lisosomi; la ferroportina
controlla a sua volta l'assorbimento intestinale di ferro e il
corretto trasporto del metallo nel macrofago. L'attività
dell'epcidina è regolata da una serina proteasi
transmembrana, codificata dal gene TMPRSS6 (cromosoma 22), che
risulta mutato in questa condizione patologica.
Luca
giunge alla nostra osservazione all'età 13 anni per anemia
sideropenica cronica.
All'età
di cinque anni un esame emocromocitometrico occasionale evidenzia la
presenza di anemia microcitica (Hb: 8 g/dl, MCV:55 fl), con conta
reticolocitaria 45000/ul, ferritina 7,3 ng/ml e sideremia 10 µg/dl.
Viene avviata supplementazione marziale per via orale senza
miglioramento significativo dei parametri ematologici (Hb 8,7 g/dl,
HCT 31%, MCV 57 fl, ferritina 59 ng/ml, sideremia 16 ug/dl,
transferrina 233 mg/dl, saturazione transferrina 4%). Viene per tale
motivo sottoposto a numerosi accertamenti.
Per
escludere un'emoglobinopatia, quale concausa della microcitosi,
viene eseguita l'elettroforesi dell'emoglobina risultata nella
norma.
Vengono
avviate indagini volte allo studio del tratto gastrointestinale
nell'ipotesi di una perdita cronica (ricerca di sangue occulto
fecale, antigene fecale per Helicobacter pylori e Urea Breath
Test, colonscopia, esofagogastroduodenoscopia (EGDS) e scintigrafia
intestinale per escludere un diverticolo di Meckel) e di un
malassorbimento (tra gli altri screening per la malattia celiaca,
coproculture e ricerche parassitologiche nulle feci). In occasione
della prima EGDS si dimostra un'infezione da Giardia lamblia
e si avvia un ciclo eradicante con metronidazolo.
Nel
sospetto di un'alterazione congenita dell'emopoiesi viene
sottoposto a osteomielobiopsia che mostra normale cellularità
e maturazione emopoietica.
Per il
persistere dell'anemia viene sottoposto a una seconda EGDS, che non
risulta dirimente, al pari di tutte le altre indagini.
Dopo
alcuni cicli di supplementazione marziale per via orale, sempre
inefficaci, esegue un trattamento con ferro per via parenterale (90
mg di gluconato ferro sodico a cadenza settimanale per 5 settimane)
con un discreto risultato: l'emoglobina prima del trattamento è
pari a 9,7 g/dl (MCV 62,2 fl), l'emoglobina alla fine del
trattamento risulta pari a 11,3 g/dl (MCV 69 fl).
Quando
giunge alla nostra osservazione, il ragazzo presenta nuovamente i
parametri classici di un'anemia sideropenica: Hb 10,6 g/dl, MCV
63,8 fl, saturazione della transferrina 6%.
Le cause
più comuni di anemia sideropenica erano già state
indagate. Pertanto, per escludere un'aceruloplasminemia viene
eseguito il dosaggio della ceruloplasmina, che risulta normale; il
valore elevato di transferrina (312 mg/dl) permette di escludere
un'ipotransferrinemia.
Prima di
effettuare un nuovo tentativo terapeutico con ferro per via
parenterale, si ritiene indispensabile escludere una malattia da
accumulo di ferro. La normalità della spirometria e della
biosuscettometria magnetica SQUID esclude con ragionevole certezza un
accumulo polmonare (emosiderosi polmonare) e un accumulo epatico,
rispettivamente.
Si
ripristina, dunque, il trattamento con ferro sucralfato per via
parenterale (100 mg/settimana x 8 settimane) ottenendo un discreto
incremento del livello di emoglobina (11,4 g/dl). Purtroppo, Luca
ritorna alla nostra attenzione dopo 6 mesi, ancora una volta anemico.
Esclusa
nuovamente l'ipotesi di anemia da perdite gastrointestinali, ci si
orienta verso la ricerca di alterazioni a carico delle proteine
coinvolte nel metabolismo del ferro, in particolare DMT1 e TMPRSS6.
L'analisi molecolare evidenzia così una mutazione di TMPRSS6
(V736A in omozigosi), permettendo la diagnosi di IRIDA (Iron
Refractory Iron Deficiency Anemia). I genitori risultano
eterozigoti per la medesima mutazione.
L'anemia
microcitica è la forma di anemia più comune dell'età
pediatrica. Comprende un gruppo molto eterogeneo di patologie che
possono sia acquisite che dovute a un difetto congenito.
La
presenza di globuli rossi con volume corpuscolare medio (MCV) ridotto
è dovuta generalmente a una ridotta sintesi di emoglobina.
Essa può essere ricondotta sia a difetti di assorbimento o di
disponibilità di ferro, sia a difetti della sintesi globinica
(emoglobinopatie) o del gruppo eme.
Tra le
forme acquisite, la causa più frequente è la carenza di
ferro; secondo le statistiche del WHO, il 43% dei bambini di tutto il
mondo sono sideropenici1 e nei paesi industrializzati il
17% dei bambini sotto i 5 anni soffre di anemia sideropenica2.
La
carenza di ferro è definita da una saturazione di transferrina
<10% e da valori di ferritina <12 mg/l3. Le cause
della carenza di ferro sono l'apporto insufficiente, il
malassorbimento intestinale, la perdita da sanguinamento e
l'aumentato fabbisogno (Tabella I).

La causa
più frequente di anemia sideropenica in età pediatrica
è l'inadeguato apporto marziale da svezzamento tardivo4.
Secondo le linee guida WHO nei neonati sani, nati a termine di
gravidanza, l'allattamento materno esclusivo deve essere mantenuto
fino al sesto mese di vita; l'Accademia Americana di Pediatria
(AAP) lo raccomanda almeno per quattro mesi ma comunque
preferibilmente fino a sei mesi. Un neonato normale in
allattamento materno esclusivo ha riserve di ferro solo sino al
raddoppio del peso neonatale: le scorte fisiologiche si esauriscono,
infatti, tra il quarto e il sesto mese di vita. L'allattamento al
seno se viene mantenuto esclusivo oltre il sesto mese di vita è
associato a un aumentato rischio di anemia sideropenica a nove mesi
di vita5.
Durante
l'infanzia il ferro è necessario sia per l'espansione
della massa eritrocitaria che per l'accrescimento
staturo-ponderale, nell'adulto invece il ferro è utilizzato
esclusivamente per l'emopoiesi; questo spiega la maggiore gravità
dell'anemia nell'età infantile rispetto all'età
adulta.
Proseguendo
nella disamina diagnostica, occorre indagare i casi da causa non
chiarita e/o non responsivi al trattamento marziale per alterazione
di proteine implicate nel metabolismo del ferro dovute a un difetto
genetico.
Per
identificare i geni potenzialmente mutati nell'anemia ipocromica
microcitica, si propone la Figura
16
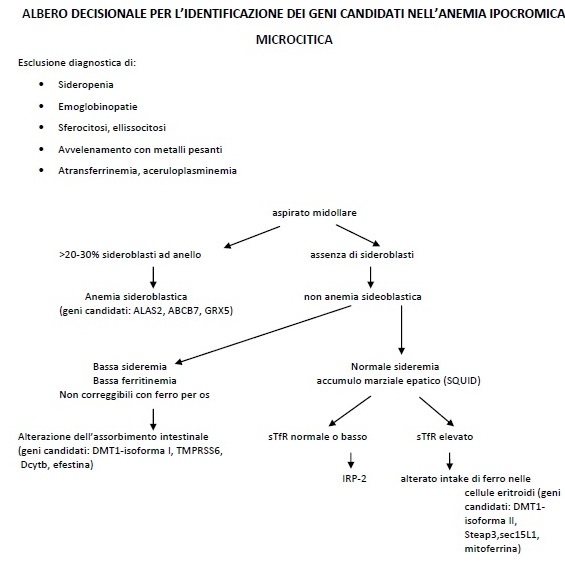
Figura
1. Albero decisionale per l'identificazione dei geni
candidati nell'anemia ipocromica. Adattato da Iolascon, et al.
20096.
Nel caso
di Luca, numerosi test diagnostici hanno escluso cause acquisite di
carenza marziale quali perdite ematiche gastroenteriche, malattie
infiammatorie croniche, sindromi malassorbitive e cause ereditarie di
microcitosi.
Così
com'è avvenuto per Luca, i casi di anemia sideropenica non
responsivi al trattamento di ferro per os e solo parzialmente al
ferro parenterale devono essere indagati per IRIDA.
L'IRIDA
è un'anemia da alterato assorbimento e utilizzo di ferro7,
dovuta a una mutazione del gene TMPRSS6, presente sul cromosoma 22,
codificante per la proteina matriptasi-2 (serina proteasi
transmembrana) prodotta dal fegato, la quale inibisce l'espressione
di epcidina.
L'epcidina
è un peptide prodotto anch'esso dal fegato, che controlla il
metabolismo del ferro inducendo la degradazione della ferroportina,
la proteina responsabile del rilascio del ferro dalla cellula al
sangue. La ferroportina, presente sia sulla membrana basale
dell'enterocita che sulla membrana macrofagica controlla
l'assorbimento intestinale di ferro e il corretto trasporto del
metallo a livello macrofagico (Figura 2).
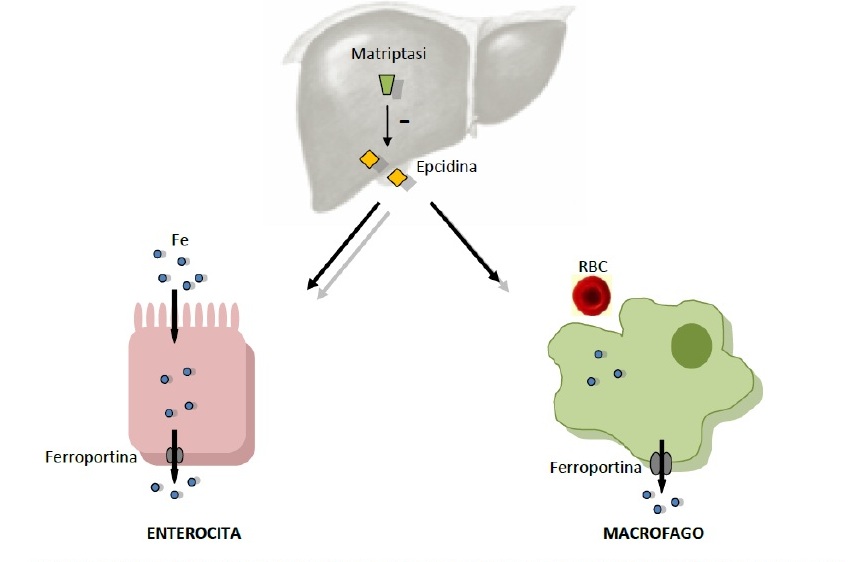
Figura
2. Situazione fisiologica: attraverso l'enzima matriptasi,
il fegato regola la sintesi di epcidina: man mano che il ferro si
riduce, nell'organismo viene inibita la sintesi di epcidina,
aumenta la ferroportina ed elevate quantità di ferro
transitano dall'intestino al lume vascolare attraverso l'enterocita
(si apre la porta del ferro).
Nell'IRIDA
la mutazione di TMPRSS6 comporta un aumento dell'espressione di
epcidina che, inibendo la ferroportina sia livello macrofagico sia a
livello enterocitario, determina un mancato assorbimento intestinale
di ferro e il suo alterato trasporto all'interno del macrofago
(Figura 3).
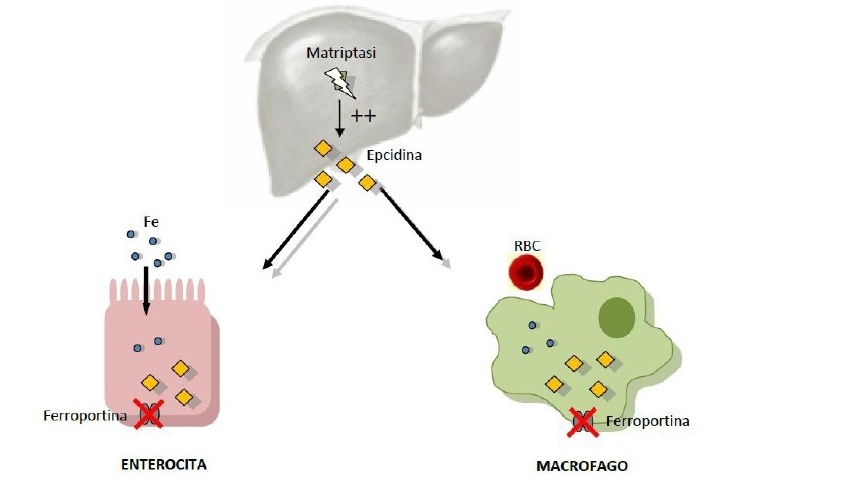
Figura
3. IRIDA. La matriptasi mutata non controlla più la
sintesi di epcidina. Il suo livello aumenta anche in carenza di
ferro, la ferroportina viene degradata e il transito del ferro è
impedito.
Il quadro
clinico dell'IRIDA è quello classico dell'anemia
sideropenica: anemia ipocromica, volume corpuscolare medio
eritrocitario inferiore ai limiti di norma e bassa saturazione di
transferrina. È caratterizzata, inoltre, da alterato
assorbimento di ferro (come evidenziato dal mancato miglioramento dei
parametri ematologici in seguito al trattamento con ferro orale),
alterato utilizzo di ferro (come evidenziato dalla risposta lenta e
incompleta al ferro parenterale) e un pattern ereditario compatibile
con una trasmissione autosomica recessiva.
Il caso
di Luca evidenzia che talora l'IRIDA non presenta solo deficit di
assorbimento intestinale, ma anche alterato metabolismo del ferro,
con la possibilità di ripresentare anemia anche in assenza di
perdita.
1. Afzal
M, Qureshi SM, Lutafullah M, Iqbal M, Sultan M, Khan SA. Comparative
study of efficacy, tolerability and compliance of oral iron
preparations (iron edetae, iron polymatose complex) and intramuscular
iron sorbitol in iron deficiency anaemia in children. J Pak Med Assoc
2009;59:764-8.
2. Pinsk
V, Levy J, Moser A, Yerushalmi B, Kapelushnik J. Efficacy and safety
of intravenous iron sucrose therapy in a group of children with iron
deficiency anemia. Isr Med Assoc J 2008;10:335-8.
3. Huang
SC, Yang YJ, Cheng CN, Chen JS, Lin CH. The etiology and treatment
outcome of iron deficiency and iron deficiency anemia in children. J
Pediatr Hematol Oncol 2010;32:282-5.
4.
Dallman PR. Nutritional anemias in childhood: iron, folate and
vitamin B12. In: Suskind RM, Lewinter-Suskind L eds. Textbook of
Pediatric Nutrition. 2nd ed. New York, NY: Raven Press 1993:91-105.
5. Baker
RD, Greer FR; Committee on Nutrition American Academy of Pediatrics.
Diagnosis and prevention of iron deficiency and iron-deficiency
anemia in infants and young children (0-3 years of age). Pediatrics
2010;126;1040-50.
6.
Iolascon A, De Falco L, Beaumont C. Molecular basis of inherited
microcytic anemia due to defects in iron acquisition or heme
synthesis. Haematologica 2009;94:395-408.
7.
Finberg KE. Iron-refractory iron deficiency anemia. Semin Hematol
2009;46:378-86.
Vuoi citare questo contributo?