Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Gennaio 2002 - Volume V - numero 1
M&B Pagine Elettroniche
Pediatria per l'ospedale
Febbre
periodica ereditaria
(Parte
prima)
Negli
ultimi anni è sorto un grande interesse da parte dei pediatri
sul problema della febbre periodica. Ne sono testimonianza le
numerose lettere su Pediatria on Line (vedi un caso descritto dal
dottor Tripodi di Reggio Calabria, di cui è stato parlato su
FORUM) e i numerosi interventi su Medico e Bambino (Tommasini A,
Lepore L 18:506-10, 1999; Tommasini A, Neri E 20:225-9, 2001; D'Agaro
P, Panizon F, Ventura A, Zocconi E 20:231-4, 2001; Besoli G e Gruppo
di studio sulla PFAFA del Friuli-Venezia Giulia 20:234-8, 2001).
La
pubblicazione, sotto la rubrica "Medical Progress"del New
England Journal of Medicine (Drenth JPH, van der Meer JWM.
Hereditary Periodic Fever. NEJM 2001, 345:1748:57), di una
revisione delle forme ereditarie della febbre periodica offre lo
spunto per ritornare sull'argomento.
Definizione
Viene
indicata con il nome di febbre periodica una febbre che presenta
delle ricorrenze, della durata da pochi giorni a poche settimane,
separate da un intervallo senza sintomi, di varia durata.
Questo
andamento della febbre può essere dovuto a diverse cause: da
forme infettive ricorrenti o malattie neoplastiche ma anche ad
alterazioni infiammatorie non infettive. A ben guardare i pazienti
che hanno febbre periodica che persiste da più di due anni è
difficile che abbiano una malattia infettiva o un tumore maligno.
D'altra
parte il susseguirsi di attacchi, con decorso sempre uguale, meglio
se in una famiglia che ha qualche componente con attacchi simili, può
suggerire la presenza di una forma non infettiva di febbre periodica.
Per esempio l'artrite cronica giovanile, o la malattia di Still a
inizio nell'adulto, o il morbo di Crohn o la sindrome di Behçet,
possono causare febbre periodica, come alcune situationi patologiche
su base ereditaria. E' proprio di queste forme che verrà in
seguito parlato:
" La
febbre mediterranea familiare
" La
sindrome da iper-IgD
" La
sindrome periodica associata al recettore del tumor necrosis factor
(TNF)
La
febbre mediterranea familiare
La
malattia è caratterizzata da brevi attacchi di sierosite
(peritonite, pleurite, artrite), spesso associata a esantema e sempre
a febbre. Il 90% dei pazienti ha il primo attacco prima dei 20 anni
di età. Le crisi sono improvvise e durano poco (da 6 a 96 ore)
(Vedi Figura n.1).
Il dolore
addominale è un'evenienza quasi costante (95% dei pazienti):
esso mima l'appendicite, la colecistite o la peritonite. Molti di
questi bambini infatti hanno subito, senza successo, interventi di
vario tipo, ora a carico di una sede anatomica, ora di un'altra.
Altre volte il quadro è dominato dalla comparsa di una
monoartrite con versamento (75% dei pazienti), al ginocchio, alla
caviglia o al polso. E' rara la presenza di un'artrite migrante o di
un'artrite cronica distruttiva. Il dolore toracico unilaterale si
ritrova nel 30% dei pazienti e la pericardite in meno dell'1%.
Un
esantema sulle gambe o sui piedi, simile all'eresipela, è
presente nel 7-40% dei pazienti: esso compare con la febbre e
sparisce appena essa sia scomparsa.
Rare sono
le mialgie gravi e persistenti delle gambe.
La
principale complicazione della malattia è l'amiloidosi,
soprattutto a carico del rene, ma anche dell'intestino, del fegato,
della milza o del cuore, dei testicoli o della tiroide. L'amiloidosi
è frequente negli ebrei Sefardici e più rara negli
ebrei Ashkenazi.
Genetica
ed epidemiologia
La febbre
mediterranea familiare è una malattia autosomica recessiva: è
la più frequente delle febbri periodiche, perché
colpisce circa 10.000 persone in tutto il mondo, soprattutto nel
bacino del Mediterraneo (ebrei Sefardici, Arabi, Turchi e Armeni).
Pur essendo la malattia rara in altre popolazioni, qualche caso è
stato anche descritto fra i Greci, gli Italiani, i Cubani e i Belgi.
La frequenza del gene delle malattia varia da un popolo all'altro:
fra gli Armeni il rapporto fra una persona portatrice del gene e
quelle che non lo sono è di 1 a 7, fra gli ebrei Sefardici da
1:5 a 1:16 e fra gli ebrei Ashkenazi di 1:135.
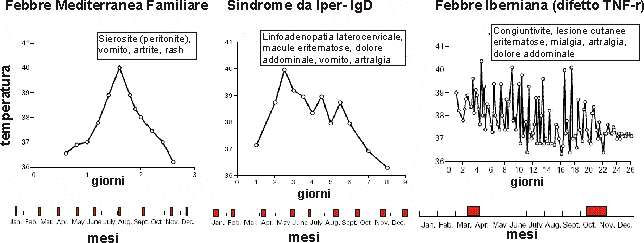
Figura
n. 1 - Andamento di un episodio tipico di febbre (tracciato
superiore), e della ricorrenza degli episodi in un anno (barra
inferiore) nelle diverse sindormi. Da N Engl J Med, adattata.
Ogni
grafico mostra la temperatura durante l'attacco di febbre e i sintomi
che si accompagnano alla febbre. La barra al di sotto del grafico,
suddivisa in mesi mostra il numero degli attacchi che il paziente ha
in un anno e la loro durata approssimativa. Il paziente con la
sindrome periodica associata al recettore del TNF ha attacchi
prolungati due volte all'anno, mentre il paziente con febbre
mediterranea familiare e il paziente con sindrome da IgD hanno
attacchi più corti ma più frequenti.
Esami
di laboratorio
Non c'è
un esame specifico per la malattia. E' vero che i pazienti colpiti
mancano di una proteasi specifica, normalmente presente nei liquidi
delle sierose, che inattiva sia l'interleuchina 8 che l'inibitore del
fattore 5a chemiotattico del complemento, ma la ricerca di questa
proteina non è possibile nella routine clinica.
Fra i
reperti non specifici vanno ricordati alcuni mediatori della flogosi,
come l'amiloide A del siero, il fibrinogeno e la proteina C reattiva,
che si innalzano durante gli attacchi febbrili. La presenza di
proteinuria (oltre 0.5 g di proteinuria nelle 24 ore) è
indicativa di amiloidosi renale.
Patogenesi
ll gene
della malattia (MEFV) è stato localizzato sul cromosoma 16. La
proteina (pirina o marenostrin) è costituita da 781 aminoacidi
e ha un peso molecolare di 86.000 daltoni. Il gene MEFV è
espresso principalmente nelle cellule mieloidi; i mediatori
dell'infiammazione, come l'interferon- e il tumor necrosis factor
sono stimoli efficaci per attivare l'espressione del gene. La precisa
funzione della pirina non è ancora chiara: essa è
espressa principalmente nel citoplasma dei neutrofili e dei monociti
maturi e si pensa sia necessaria per regolare l'infiammazione mediata
dai neutrofili.
Sono
state individuate almeno 28 mutazioni a carico del gene MEFV, la
maggior parte delle quali concentrate in un esone (il numero 10). Due
mutazioni missense sono la M694V (ritrovata nel 20-67% dei casi) e la
V726A (ritrovata nel 7-35% dei casi): la loro prevalenza varia con la
popolazione studiata. Viene ritenuto che le due mutazioni avvennero
in un comune antenato, vissuto circa 2.500 anni fa, nel medio
oriente. E' stato ipotizzato, per l'alta frequenza della mutazione,
che il portatore di queste mutazioni possa presentare una maggiore
resistenza per un patogeno che non è stato ancora
identificato.
La
mutazione M694V si accompagna a un fenotipo più grave e più
facilmente portato all'amiloidosi.
Trattamento
La
colchicina rappresenta il farmaco di scelta nei pazienti con febbre
mediterranea familiare: previene gli attacchi febbrili nel 60% dei
pazienti e ne riduce il numero in un altro 20-30%. Non risponde il
5-10% dei pazienti, ma è stato pensato che questo possa essere
dovuto a una mancanza di compliance.
La dose
media per un soggetto adulto è di 1 mg al giorno, ma se la
malattia non risponde è possibile dare anche 2 o 3 mg al
giorno. La maggioranza dei pazienti tollera queste dosi, ma se
insorgesse diarrea e dolori addominali è necessario ridurle.
Fra gli effetti collaterali sono da ricordare anche la miopatia, la
neuropatia e la leucopenia in soggetti con interessamento epatico o
renale. La colcichina non arresta un attacco quando questo sia già
iniziato. Il diclofenac può essere usato alla dose di 75 mg
per intramuscolo in un adulto per calmare il dolore.
La
sospensione della colchicina può accompagnarsi alla comparsa
di un attacco entro pochi giorni.
L'uso di
altri farmaci non ha retto a un controllo approfondito
dell'efficacia.
Prognosi
La
prognosi è collegata strettamente alla presenza o meno di
amiloidosi. Quando essa non ci sia, la prognosi quod vitam è
buona. Prima dell'uso della colchicina l'amiloidosi insorgeva nel 60%
dei pazienti che avevano superato i 40 anni di età. Essa
rappresentava la causa più frequente di morte. Anche se la
colchicina non riesce a prevenire gli attacchi di febbre, essa
comunque impedisce lo sviluppo dell'amiloidosi.
Strategia
diagnostica
Per prima
cosa è essenziale una precisa raccolta dell'anamnesi. In
secondo luogo sarebbe bene vedere il paziente durante un attacco.
Se il
paziente ha la storia clinica caratteristica e appartiene a un gruppo
etnico, nel quale sia presente un'alta prevalenza della malattia,
porre la diagnosi non è difficile (Vedi Tabella n. 1). Certo,
per porre la diagnosi, bisogna avere prima di tutto la conoscenza
dell'esistenza della malattia e la capacità (elemento più
difficile) di applicare nella pratica, la propria cultura.
Poiché
il gene MEFV è stato clonato, è possibile ottenere una
diagnosi molecolare della febbre mediterranea familiare, ma a questo
proposito esistono alcune limitazioni. I laboratori ricercano le 5
mutazioni che si riscontrano più di frequente (M694V, V726A,
V680I, E148Q e V694I e non ricercano le altre mutazioni più
rare, per cui può risultare negativo anche un soggetto che in
effetti sia colpito dalla malattia.
Inoltre,
le mutazioni MEFV avvengono in ambedue gli alleli nel 70% dei casi
tipici; nel rimanente 30% può esserci la mutazione in un solo
allele o più di rado in nessuno. Tuttavia nonostante queste
limitazioni la prova molecolare può essere usata come una
prova di conferma nei casi in cui ci sia un elevato grado di
sospetto. Se il quadro è molto sospetto, anche se le prove
molecolari sono negative, va ugualmente usato il trattamento con
colchicina.
Tabella
n. 1- Aspetti distintivi fra le 3 forme di febbre periodica
ereditaria
#B#Aspetti | Febbre
mediterranea familiare | Sindrome
da Iper-IgD | S.
periodica associata al recettore del TNF |
Origine | Ebrea,
Turca | Olandese,
Francese | Scozzese,
Irlandese |
Trasmissione
familiare | Orizzontale* | Orizzontale* | Verticale* |
Età
all'inizio in anni | <
20 anni | <
1 anno | <
20 anni |
Durata
tipica dell'attacco in giorni | <
2 | 4-6 | >
14 |
Sintomi,
oltre la febbre | Sierosità,
interessamento dello scroto, eritema | Linfoadenopatia
cervicale | Congiuntivite,
mialgia , localizzata |
Reperti
di laboratorio | Bassi
livelli di inibitore del C5 nei liquidi delle sierose | IgD
seiriche elevate (>100 UI/mL) | Bassi
livelli sierici del recettore del TNF tipo 1 (< 1 ng/mL) |
Gene | MEFV | Gene
della mevalonato chinasi | Gene
del recettore del TNF tipo 1 |
Proteina | Pirina | Mevalonato
chinasi | Recettore
del TNF tipo 1 |
Terapia | Colchicina | Cure
non disponibili | Corticosteroidi,
etanercept |
Vuoi citare questo contributo?