Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Ottobre 2005 - Volume VIII - numero 8
M&B Pagine Elettroniche
Protocolli di diagnosi e terapia
La
tirosinemia epatorenale
Centro di
Malattie Metaboliche Clinica Pediatrica, Università di Torino
Indirizzo
per la corrispondenza: redazione@medicoebambino.com
La
tirosinemia epatorenale (tirosinemia di tipo I) è una malattia
ereditaria del catabolismo della tirosina, responsabile di una severa
epatopatia, di una tubulopatia renale generalizzata e di una
polineuropatia periferica acuta di tipo porfirico.
Le prime
descrizioni cliniche della malattia risalgono agli inizi degli anni
sessanta1,
ma solo nel 1977, grazie alla dimostrazione di elevate concentrazioni
urinarie di succinilacetone (SA) nei pazienti, è stato
possibile identificare il difetto biochimico a livello dell'ultimo
enzima della degradazione della tirosina, la fumarilacetoacetato
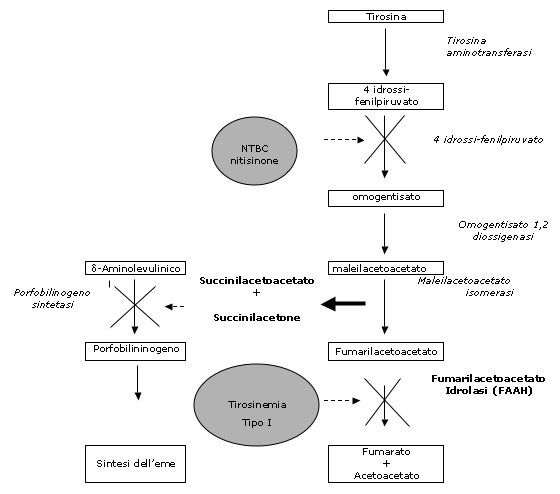
idrolasi (FAAH)2
(Fig.1).
La malattia riconosce una trasmissione di tipo autosomico recessivo
ed il gene che codifica per la FAAH è stato localizzato sul
cromosoma 15 (15q23-q25)3.
FISIOPATOLOGIA
Il
deficit di FAAH causa l'accumulo di fumarilacetoacetato (FAA), di
maleilacetoacetato (MAA) e dei loro derivati succinilacetoacetato
(SAA) e succinilacetone (SA) (Fig.1).
Il FAA ed
il MAA sono dei composti alchilanti, responsabili di un effetto
tossico a livello del fegato e dei tubuli renali e, molto
probabilmente per le loro proprietà mutagene, dello sviluppo
della neoplasia epatica4.
La struttura del MAA è simile a quella dell'acido maleico,
che viene utilizzato negli studi sperimentali delle tubulopatie.
Inoltre, è stato anche dimostrato che l'SA ad alte
concentrazioni è in grado di causare l'inibizione del
trasporto tubulare del glucosio, degli aminoacidi e dei fosfati5,6.
La patogenesi delle crisi neurologiche di tipo porfirico è
invece da porsi in relazione con l'accumulo dell'acido
d-aminolevulinico secondario al blocco dell'enzima porfobilinogeno
sintasi da parte dell' SA (Fig.1).
MANIFESTAZIONI
CLINICHE
Fegato
La
tirosinemia di tipo I è una malattia essenzialmente epatica.
Nella maggior parte dei pazienti, la presentazione della
sintomatologia avviene nel corso delle prime settimane o mesi di vita
(normalmente entro il sesto mese) nel contesto di una severa
insufficienza epatica, con sindrome emorragica, ipoglicemia, ittero,
edema, ascite, epatosplenomegalia.
Un gruppo
di pazienti (circa il 20 %) giunge alla attenzione medica in epoca
più tardiva, in quanto la malattia epatica può
presentarsi anche in una forma cronica con segni clinici più
aspecifici (epatomegalia isolata, ipertransaminasemia moderata,
vomito cronico, anoressia, scarsa crescita) che possono ritardare la
diagnosi.
E'
molto importante sottolineare che tutti i pazienti, sia quelli con
esordio acuto che quelli con forma cronica, sviluppano una
degenerazione cirrotica (micro/macronodulare) e il rischio di
insorgenza di un epatocarcinoma precoce è elevatissimo.
Rene
La
tubulopatia rappresenta la manifestazione nefrologica principale
della tirosinemia. I pazienti in genere presentano una disfunzione
tubulare di tipo generalizzato (sindrome di Fanconi con glicosuria,
iperaminoaciduria, fosfaturia).
A livello
istologico, lesioni tubulo-interstiziali (dilatazioni tubulari con
vacuolizzazione delle cellule epiteliali, fibrosi interstiziale)
possono anche associarsi ad una sclerosi glomerulare7.
La perdita urinaria dei fosfati è all'origine del rachitismo
ipofosfatemico vitamino-resistente, che rappresenta una delle
complicazioni più frequenti della malattia. In certi casi, la
sindrome di Fanconi diviene irreversibile e costituisce il problema
medico principale8.
All'esame ecografico si trova frequentemente una nefromegalia, che
può essere associata a nefrocalcinosi9.
Certi pazienti presentano anomalie della fitrazione glomerulare e
possono andare incontro ad insufficienza renale.10
La caratterizzazione del danno renale è molto importante per
quei pazienti che andranno eventualmente incontro a trapianto (scelta
fra trapianto epatico isolato o trapianto combinato fegato-rene).
Nervi
periferici
L'inibizione
del metabolismo porfirinico con conseguente aumento delle
concentrazioni di acido d-aminolevulinico, secondario all'accumulo
di SA, è alla base degli episodi di polineuropatia periferica
acuta che si manifesta con parestesie dolorose e paralisi
progressiva. Tutti i bambini tirosinemici sono a rischio di tali
crisi neurologiche, spesso scatenate da infezioni intercorrenti, e
che possono condurre ad insufficienza respiratoria acuta da paralisi
muscolare. L'esame istologico del nervo mostra una degenerazione
assonale con demielinizzazione secondaria 11
.
CLASSIFICAZIONE
CLINICA
E'
stata proposta una classificazione della tirosinemia in tre forme
cliniche principali: una forma acuta che si manifesta prima dei sei
mesi di vita con un coinvolgimento epatico severo e a volte
fulminante; una forma sub-acuta che si palesa tra i 6 mesi e l'anno
di vita con epatomegalia, ritardo della crescita e rachitismo; e una
forma cronica che si rivela dopo i 12 mesi, caratterizzata
soprattutto dalle alterazioni scheletriche12.
Più
recentemente uno studio internazionale di tipo retrospettivo ha
rivisitato questa classificazione e, utilizzando un criterio
prognostico, ha suggerito di suddividere la forma acuta in due
gruppi: una forma molto precoce (very early form) che si rivela prima
dei due mesi, ed una forma precoce (early form) che esordisce tra i 2
ed i 6 mesi di vita; tutti i pazienti che manifestano i primi sintomi
dopo i 6 mesi di vita sono stati invece classificati in un'unica
forma cronica e tardiva (late form)13.
DIAGNOSI
METABOLICA
In ogni
paziente con sospetto clinico di tirosinemia epatorenale, deve essere
richiesta l'analisi degli acidi organici urinari al fine di
evidenziare una elevata escrezione di succinilacetone. Benchè
questo composto rappresenti il marker specifico per la diagnosi
formale di tirosinemia, a scopo diagnostico è sempre molto
utile effettuare anche il dosaggio urinario dell'acido
d-aminolevulinico, test dotato di altissima sensibiltà
diagnostica poiché questo metabolita è sempre presente
in concentrazioni elevate nei pazienti tirosinemici.
La
dimostrazione di elevate concentrazioni urinarie di succilacetone e
di acido d-aminolevulinico è sufficiente per affermare la
diagnosi. L'analisi degli aminoacidi plasmatici non rappresenta
invece un valido aiuto diagnostico, in quanto l' ipertirosinemia
non è patognomonica per questa malattia, ma può essere
osservata in tutte le cause di insufficienza epatocellulare, spesso
in associazione con l'iperfenilalaninemia e l'ipermetioninemia.
Inoltre, in alcuni casi di tirosinemia epatorenale le concentrazioni
plasmatiche di tirosina possono essere del tutto normali.
L'analisi
degli aminoacidi urinari può essere utile al fine di mettere
in evidenza l'iperaminoaciduria generalizzata.
Infine,
la maggioranza dei pazienti presenta al momento della diagnosi
elevati valori plasmatici di a-feto-proteina.
EPIDEMIOLOGIA
In alcune
regioni sno stati avviati degli studi pilota di screening neonatale
di massa della malattia che hanno dimostrato che l' incidenza della
tirosinemia epatorenale è di 1 caso ogni 100.000 nascite.
Nella regione canadese del Quebec lo screening neonatale della
tirosinemia è divenuto pratica corrente da molti anni poiché
in questa zona si è osservata una frequenza particolarmente
elevata (1:2000 nascite).
TRATTAMENTO
La
riduzione o l'abolizione della produzione di SA e degli altri
metaboliti tossici rappresenta il target di ogni strategia
terapeutica della tirosinemia epatorenale.
La dieta
controllata negli apporti di fenilalanina e tirosina rappresenta da
anni la base del trattamento della malattia14.
L'approccio dietetico è fondamentale nelle fasi acute, ma
non è sufficiente per arrestare la progressione delle
complicanze croniche e tumorali della malattia, né per
prevenire il rischio di crisi neurologiche e di decompensazioni
epatiche13.
Dal 1978,
il trapianto epatico offre una alternativa terapeutica efficace per i
bambini tirosinemici15.
Il trapianto previene le manifestazioni epatiche acute, il rischio di
epatocarcinoma e la comparsa de polineuropatie periferiche. Ciò
nonostante, la prognosi renale dopo trapianto epatico non è
certissima, in quanto la produzione endogena renale dei metaboliti
tossici, anchè se molto inferiore rispetto a quella epatica,
persiste16
e non si può escludere a priori l'eventualità di una
nefropatia nel corso degli anni
17. Nei pazienti con danno renale severo è stato
anche effettuato un trapianto combinato 9.
All'inizio
degli anni 1990 si è assistito ad un radicale cambiamento
nell'approccio terapeutico della tirosinemia epatorenale. Un gruppo
svedese ha proposto la terapia farmacologica con 2-(2
nitro-4-trifluorometilbenoil)-1,3 cicloesandione (NTBC)18,
composto derivante da ricerche effettuate nell'ambito dell'
agronomia19.
Questa molecola si è dimostrata in grado di inibire il secondo
enzima della degradazione della tirosina, la 4-idrossi-fenilpiruvato
diossigenasi e di determinare l'abolizione completa della
produzione dei metaboliti implicati nella patogenesi della malattia.
Attualmente
la somministrazione di NTBC viene effettuata in alcune centinaia di
pazienti e i risultati presentati in un recente studio prospettico20
sono molto incoraggianti in quanto oltre il 90% ha mostrato una
risposta biologica (soppressione della produzione di SA e di acido
d-amino levulinico, normalizzazione del tasso di a-feto proteina
serica) e clinica spettacolare (normalizzazione delle funzioni
epatiche e renali, assenza di crisi porfiriche). Questo studio
dimostra, inoltre, che nessuno dei pazienti che ha iniziato la
terapia con NTBC prima dell'età di 2 anni, ha sviluppato nel
tempo l'epatocarcinoma.
Poiché
l'NTBC determina l'inibizione del catabolismo della tirosina a un
livello metabolico precoce, i pazienti in terapia presentano una
ipertirosinemia iatrogena e al fine di evitare potenziali complicanze
cutanee ed oculari, si rende necessario proseguire una dieta povera
di fenilalanina e tirosina.
CONCLUSIONI
Il nuovo
approccio terapeutico con NTBC ha determinato un mutamento radicale
nella prognosi epatica, renale e neurologica dei bambini affetti da
tirosinemia epatorenale e rappresenta ormai, dopo dieci anni di
esperienza clinica, una valida e consolidata alternativa al trapianto
epatico. Risulta quindi fondamentale individuare precocemente i
pazienti per iniziare la terapia farmacologica nelle fasi iniziali
della malattia. Poiché sulla base di una incidenza di 1:100000
nascite in Italia si attendono circa 5 nuovi casi all'anno, dal
1992 (anno in cui si è reso disponibile il farmaco) al 2002
nel nostro paese dovrebbero essere stati individuati e proposti per
il trattamento almeno una cinquantina di nuovi pazienti. Ad oggi,
però, è noto che solo 12 pazienti con tirosinemia nati
in Italia a partire dal 1992 hanno potuto usufruire di questo farmaco
salvavita (dati forniti da Orphan Europe, aggiornati ad Ottobre
2002). Questa malattia ereditaria del metabolismo appare, quindi,
largamente sottostimata in ambito clinico, mentre dovrebbe essere
posta sistematicamente in diagnostica differenziale in tutte le
epatopatie che interessano sia l'epoca neonatale che tutta l'età
pediatrica.
1. Lelong
et al.- Cirrhose congénitale et familiale, rachitisme
vitamino-résistant avec diabète phospho-gluco-aminé,
hépatome terminal. Pédiatrie 1961, 3, 221
2.
Lindblad et al.- On the enzymatic defects in hereditary tyrosinemia:.
Proc Natl Acad Sci 1977, 74, 4641
3.
Phaneuf et al.-Cloning and expression of the cDNA encoding
fumarylacetoacetate hydrolase, the enzyme deficient in hereditary
tyrosinemia: assignement of the gene to chromosome 15. Am J Hum Genet
191, 48, 525.
4.
Mitchell et al. – Hypertyrosinemia in : The Metabolic and Molecular
Basis of Inherited Disease ed n 7. Edited by Scriver et al. New York
Mc Graw Hill 1995, 1077.
5.
Spencer et al. – Effects of succinylacetone on the uptakeof sugars
and aminoacids by brush border vesicles. Kidney Int 1988, 34, 671.
6. Roth
et al. –Succinylacetone effects on renal tubular phosphate
metabolism: a model for experimental renal Fanconi syndrome. Proc Soc
Exp Biol Med 1991, 196, 428.
7. Russo
et al. – Visceral pathology of hereditary tyrosinemia type I. Am J
Hum Genet 1990, 47, 317.
8.
Fridman et al. – Visceral pathology of hereditary tyrosinemia: the
Quebec experience. Am J Hum Genet 1990,47, 338.
9.
Paradis et al.- Liver transplantation for hereditary tyrosinemia :
the Quebec experience. Am J Hum Genet 1990, 47, 338.
10.
Kvittingen et al.- Renal failure in adult patients with hereditary
tyrosinemia type I. J Inher Metab Dis 1991, 14, 53.
11.
Mitchell et al.- Neurological crisis in hereditary tyrosinemia N Engl
J Med 1990, 322, 432.
12.
Halvorsen et al.– Outcome of therapy oh hereditary tyrosinemia.
Acta Pediatr Jpn 1988, 30, 425.
13. Van
Spronsen et al.- Hereditary tyrosinemia type I: a new clinical with
difference in prognosis on dietary treatment. Hepatology 1994, 20,
1187.
14.
Halvorsen et al.– Studies on tyrosinosis: effect of low-tyrosine
and low-phenylalanine diet. Br Med J 1964, 2, 1171.
15. Fish
et al._ Persistent succinylacetone excretion after liver
transplantation in a patient with hereditary tyrosinemia type I. J
Pediatr 1978, 93, 592.
16.
Tuchman et al.- Persistent succinylacetone excretion after liver
transplantation in a patient with hereditary tyrosinemia type I. J
Inher Metab Dis 1985, 8,21.
17. Laine
et al.- The nephropathy of type I tyrosinemia after liver
transplantation. Pediatr Res 1995, 37, 640.
18.
Lindstedt et al.- Treatment of hereditary tyrosinemia type I by
inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 1992,
340,813.
19. Lock
et al.- From toxicological problem to therapeutic use: the discovery
of the mode of action of
2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), its
toxicology and development as a drug. J Inher Metab Dis 1998, 21,
498.
20. Holme
et al.- Tyrosinemia type I and NTBC
(2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J Inher
Metab Dis 1998, 21, 507.
Vuoi citare questo contributo?