Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Gennaio 2006 - Volume IX - numero 1
M&B Pagine Elettroniche
Pediatria per immagini
La
sindrome di Stevens-Johnson: antibiotico o infezione?
1 UO di
Pediatria, IRCCS Burlo Garofolo, Trieste.
2 U.O.C.
Pronto Soccorso e Primo Accoglimento, IRCSS Burlo Garofolo,
Trieste.
Indirizzo
per corrispondenza: patty76@aliceposta.it
E. è
un bambino di quasi 10 anni, la cui storia clinica inizia con una
banale infezione respiratoria caratterizzata da lieve
febbricola e tosse inizialmente stizzosa, in seguito via via più
catarrale, molto suggestiva di un quadro di bronchite,
verosimilmente virale o da Micoplasma pneumonia (MP).
Viene
avviata terapia con claritromicina. A distanza di 24 ore
dall'assunzione dell'antibiotico si osserva la comparsa di
importante edema delle labbra con evoluzione ingravescente fino
alla comparsa di lesioni ulcerative del cavo orale.
Per
questo motivo giunge in Pronto Soccorso.
Al
momento dell'arrivo in Pronto Soccorso il bambino è
piuttosto abbattuto e pallido. Ha febbricola. Presenta importante
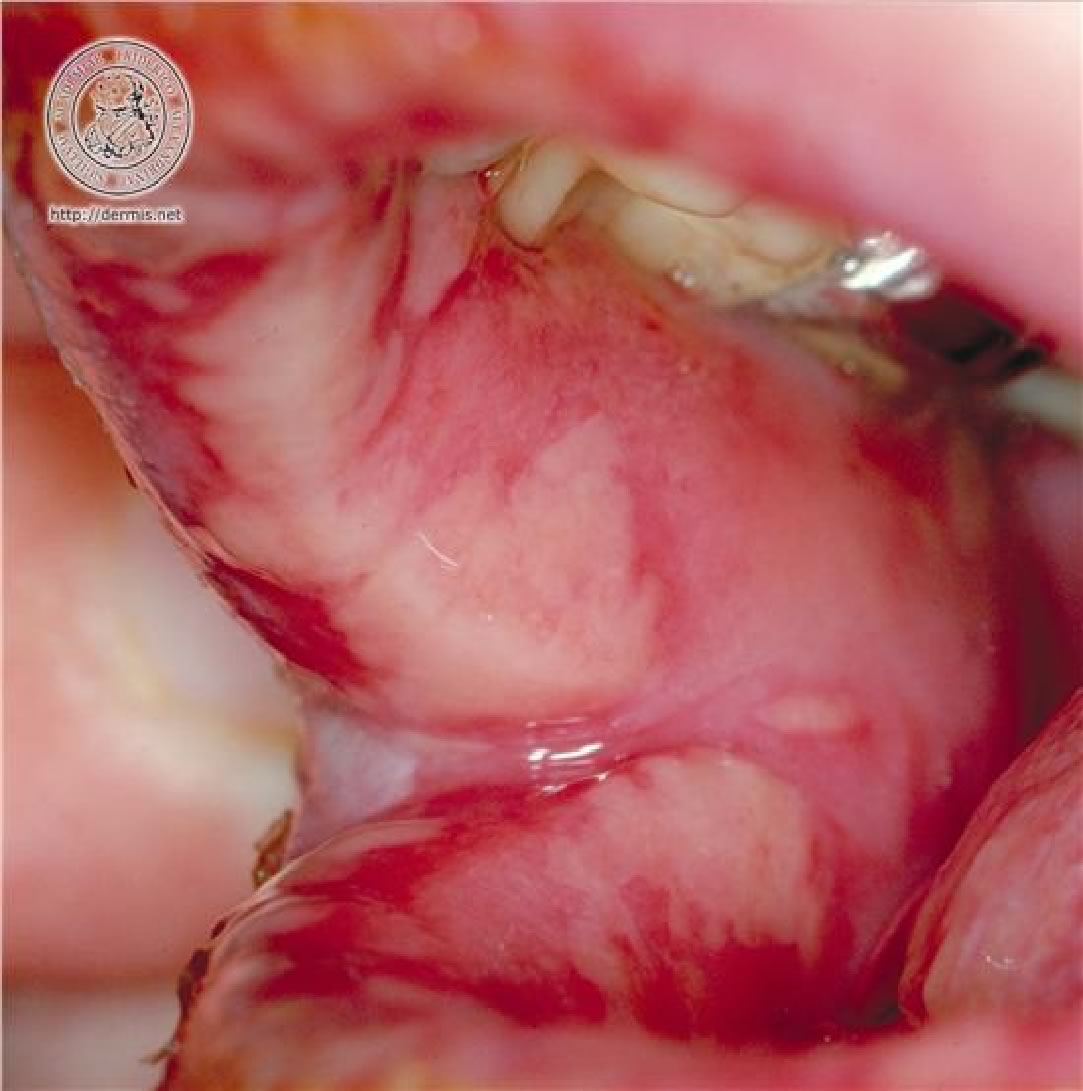
edema delle labbra con aree di disepitelizzazione. Su gengive e
palato molle si osservano numerose aree rivestite da fibrina
assieme a piccole zone ulcerate. La lingua non appare eritematosa
(Figura
1, Figura
2 a-b). Riesce ancora, anche se con fatica, a bere ed a
nutrirsi con latte e yogurt. Presenta inoltre lieve congiuntivite
non secernente.
Non
lesioni a livello cutaneo, né lesioni anali o genitali.
Non linfoadenopatia, non edema distale agli arti.
All'auscultazione
del torace si apprezzano rantoli fini crepitanti in sede basale
destra. Frequenza respiratoria nella norma. Viene eseguito un
prelievo ematico che mette in luce una lieve alterazione degli
indici di flogosi.
Il
bambino è stato quindi ricoverato. La prima ipotesi fatta
è stata quella di una sindrome di Stevens-Johnson (SJ)
frusta o atipica, vista l'assenza del coinvolgimento
cutaneo, verosimilmente causata dal MP che è uno dei
principali agenti eziologici della sindrome.
L'infezione
da MP (già ipotizzata) era suggerita dalla clinica, dalla
lunga storia con tosse inizialmente stizzosa senza un'importante
elevazione della temperatura né degli indici di flogosi, e
dal test di Krugmann francamente positivo.
In
letteratura il MP è l'agente eziologico infettivo più
frequentemente associato alla sindrome SJ. Non potevamo comunque
escludere, come causa scatenante della sindrome, un possibile
ruolo della claritromicina, farmaco a sua volta descritto in
associazione alla sindrome SJ. Dalla letteratura tuttavia sembra
necessario un periodo di latenza di almeno 3 giorni dall'inizio
dell'assunzione del farmaco perché si possa pensare ad
una eziologia iatrogena.
Pur
nel dubbio che la claritromicina non fosse responsabile della
sintomatologia, prudenzialmente il farmaco è stato sospeso
e il bambino è stato trattato con ciprofloxacina, molecola
attiva nei confronti del MP. E' stato garantito un trattamento
topico e di pulizia del cavo orale con applicazioni frequenti di
anestetici (benzocaina gel topico). Nel sospetto di una
sovrainfezione da Candida si è inoltre avviato un
trattamento con miconazolo (Daktarin oral gel).
Il
giorno successivo al ricovero si è osservata la comparsa
di importante edema dolente, inizialmente solo a livello della
guancia destra ed in seguito anche a sinistra, con rialzo
febbrile. Per tale motivo, a scopo precauzionale, nel sospetto
non escludibile di sovrainfezione da germi anaerobi (non coperti
dalla ciprofloxacina) si è aggiunta in terapia la
clindamicina, con progressivo miglioramento delle lesioni.
L'interessamento
oculare che nei primi giorni era molto lieve, si è reso
evidente in un secondo momento con coinvolgimento della cornea
(punteggiature nei settori corneali inferiori), trattate con
antibiotico locale.
La
Sindrome di Stevens-Johnson
Esiste
ancora dibattito sulla classificazione della sindrome di Steven
Johnson (SJ). La sindrome è stata inizialmente descritta
da Stevens e Johnson nel 1922 in due bambini con congiuntivite,
stomatite ed esantema generalizzato, come unentità
distinta rispetto all'eritema multiforme (EM). In realtà
negli ultimi 30 anni è abbastanza largamente accettato che
la sindrome SJ, l'EM, ed anche la epidermiolisi tossica sono
tutte parte di uno unico spettro di reazioni cutanee su base
immunologia. Sia nell'EM che nella SJ le lesioni cutanee
precoci consistono in un accumulo di cellule mononucleate attorno
ai vasi del derma.
Una
delle classificazioni proposte è la seguente:
-EM minor: caratterizzato da tipiche lesioni
eritematose a forma di bersaglio (targets) piatte o rilevate,
distribuite simmetricamente nelle regioni distali.
-EM maior: in cui le lesioni cutanee descritte per
la forma minor si associano al coinvolgimento di uno o più
superfici mucose.
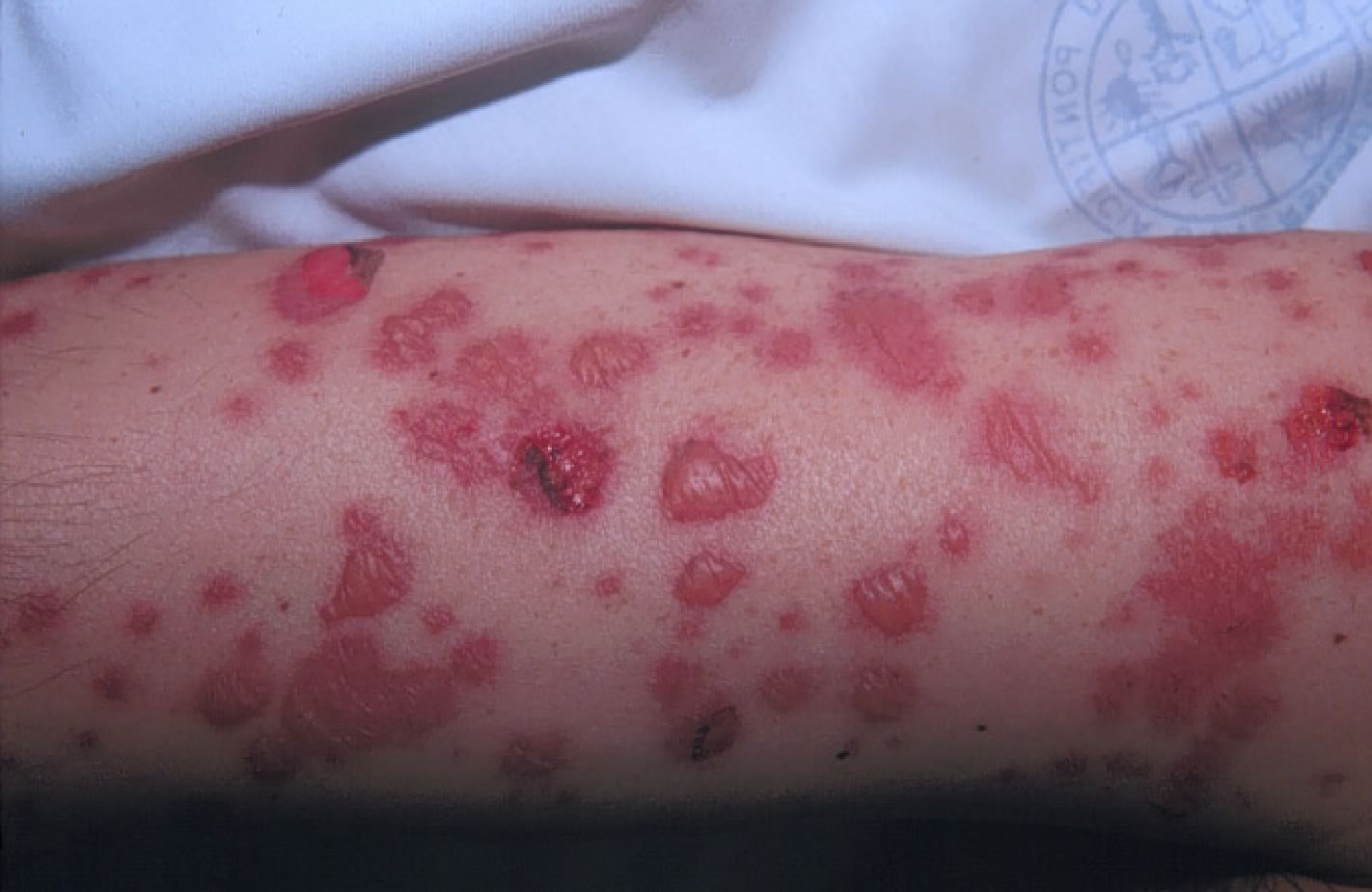
-Sindrome di SJ: caratterizzato da coinvolgimento
cutaneo con macule eritematose che sviluppano rapidamente e in
modo variabile necrosi centrale sino a formare vescicole, bolle e
aree di esfoliazione, diffuse in particolare sul tronco, volto,
ed estremità. Tipicamente le lesioni cutanee sono
accompagnate dal coinvolgimento di due o più superfici
mucose e in particolare occhi, cavità orale, alte vie
respiratorie, esofago, tratto gastrointestinale o la mucosa
anogenitale (Figure
3 a-b, Figura
4).
Comunque,
come già detto, sono possibili casi in cui si presentano
elementi caratteristiche per due forme diverse, come lesioni
target atipiche per EM, ma con distribuzione distale tipica delle
forme da EM, o come l'associazione di lesioni cutanee miste
(target tipiche, atipiche, e vescicole purpuriche), e lesioni
mucose, che rendono difficoltosa la diagnosi differenziale tra EM
maior e sindrome SJ.
Dal
punto di vista eziologico si distinguono forme associate ad
agenti infettivi e forme associate a farmaci. Il MP è
l'agente causale che con maggior certezza è stato
dimostrato essere responsabile della sindrome di SJ (circa nel
30% dei casi), mentre l'Herpes simplex risulta il principale
fattore causale dell'EM, e queste ultime forme sono soggette
spesso a ricorrenza. Tra gli altri agenti eziologici descritti lo
streptococco A, il meningococco C, e forme post vaccini vivi od
attenuati, ed un caso dopo test alla tubercolina.
Tra i
farmaci responsabili si annoverano classicamente gli antibiotici
sulfamidici, penicilline, macrolidi, gli antinfiammatori
(ibuprofene, piroxicam e i salicilati) e gli anticonvulsivanti
(fenitoina). A tal proposito, sembra che per poter riconoscere
una causa iatrogena debba intercorrere un periodo minimo di 3
giorni tra l'assunzione del farmaco e le manifestazioni
cutanee.
La
durata media della sindrome SJ è abbastanza lunga,
descritta intorno alle due settimane. Nel complesso la sindrome
SJ ha una mortalità descritta del 5%. Le complicanze più
importanti sono rappresentate dal rischio di sovrainfezione
batterica e dalle ulcerazioni corneali con conseguente
compromissione del visus. Le forme da MP sembrano associate ad un
decorso più favorevole.
La
gestione della sindrome da un punto di vista terapeutico è
sia sintomatica che di supporto. Gli anestetici per uso topico
possono dare sollievo al dolore particolarmente quando applicati
prima di mangiare. Il trattamento può richiedere il
ricovero per il supporto nutrizionale (enterale o parenterale)
nel caso in cui sia difficile l'alimentazione per bocca.
E'
doverosa una valutazione oculistica viste le possibili sequele,
quali la cicatrizzazione corneale.
La
terapia antibiotica è indicata per le sovra-infezioni
batteriche secondarie e nel caso si ipotizzi un'eziologia da
MP.
L'uso
dei corticosteroidi è molto controverso ma di regola
sconsigliato per il rischio riportato da alcuni autori di
peggiorare il quadro clinico, soprattutto se si sospetta
un'eziologia virale.
E'
descritto l'uso anedottico di Talidomide nelle forme severe o
ricorrenti, come quelle da Herpes Simplex, ma è difficile
valutarne retrospettivamente l'efficacia.
Il
caso da noi descritto rientra in una forma di SJ atipica,
in quanto deficitario delle manifestazioni cutanee
caratteristiche. Questa evenienza è descritta in
letteratura dove tra l'altro si pone l'accento su quelli che
sono i criteri imprescindibili sui quali si basa la diagnosi,
ossia la tipica stomatite, presente nel 100% dei casi associata
ad un'altra mucosite come la congiuntivite che si ritrova nel
90-100% delle forme.
Il
caso ci è parso di interesse perché ripropone le
problematiche diagnostiche, eziologiche e terapeutiche di fronte
alle quali ci si trova nei casi di sospetta o certa sindrome di
SJ.
In
particolare la storia riportata ci insegna che:
a) In
alcuni casi le manifestazioni cutanee della classica SJ possono
mancare; per poter pensare alla sindrome deve essere presente
l'interessamento del cavo orale e quello oculare, in assenza di
altri elementi clinici e di laboratorio che fanno pensare ad
altre patologie ed in primis alla malattia di Kawasaki.
b) Di
fronte ad una sindrome di SJ si pensa quasi di riflesso sempre ad
una possibile causa farmacologia; al contrario molto spesso sono
responsabili alcuni batteri (in modo particolare il MP) e virus
che innescano un meccanismo immunomediato, non ancora
completamente conosciuto nel sua patogenesi specifica. Nel
dubbio, tra infezione e farmaco, è in ogni caso importante
sospendere il trattamento in corso.
c) La
gestione è di supporto (antidolorifici, supporto
nutrizionale a volte per via parenterale), rinunciando al
cortisone (assenza di evidenze a favore; alcune segnalazioni
contro) e occupandosi delle possibili complicanze oculari.
Bibliografia
di riferimento
1.
Leaute-Labreze C, Lamireau T, Chawki D, Maleville J, Taieb
A.Diagnosis, classification, and management of erythema
multiforme and Stevens-Johnson syndrome. Arch Dis Child. 2000
;83:347-52.
2.
Vanfleteren I, Van Gysel D, De Brandt C.Stevens-Johnson syndrome:
a diagnostic challenge in the absence of skin lesions. Pediatr
Dermatol. 2003;20:52
3.
Donta-Bakoyianni K, Mitsea AG, Deodoropoulou-Papadimitriou
K.Stevens-Johnson syndrome: case presentation. J Clin Pediatr
Dent. 2002;27:71-6.
4.
Schmutz JL, Barbaud A, Trechot P.Azithromycin and Stevens-Johnson
syndrome]
Ann
Dermatol Venereol. 2005;132:728
5.
Sullivan S, Harger B, Cleary JD.Stevens-Johnson syndrome
secondary to erythromycin. Ann Pharmacother. 1999;33:1369
6.
Lam NS, Yang YH, Wang LC, Lin YT, Chiang BL.Clinical
characteristics of childhood erythema multiforme, Stevens-Johnson
syndrome and toxic epidermal necrolysis in Taiwanese children. J
Microbiol Immunol Infect. 2004 ;37:366-70.
7.
Yeung AK, Goldman RD. Use of steroids for erythema multiforme in
children.Can Fam Physician. 2005;51:1481-3.
8.
Wolf R, Orion E, Marcos B, Matz H.Life-threatening acute adverse
cutaneous drug reactions. Clin Dermatol. 2005
Mar-Apr;23(2):171-81
9.
Manders SM. Serious and life-threatening drug eruptions. Am Fam
Physician. 1995;51:1865-72. Review.
Inoltre:
Nelson
Textbook of Pediatrics. 17th Edition 2004. Saunders Company.
|
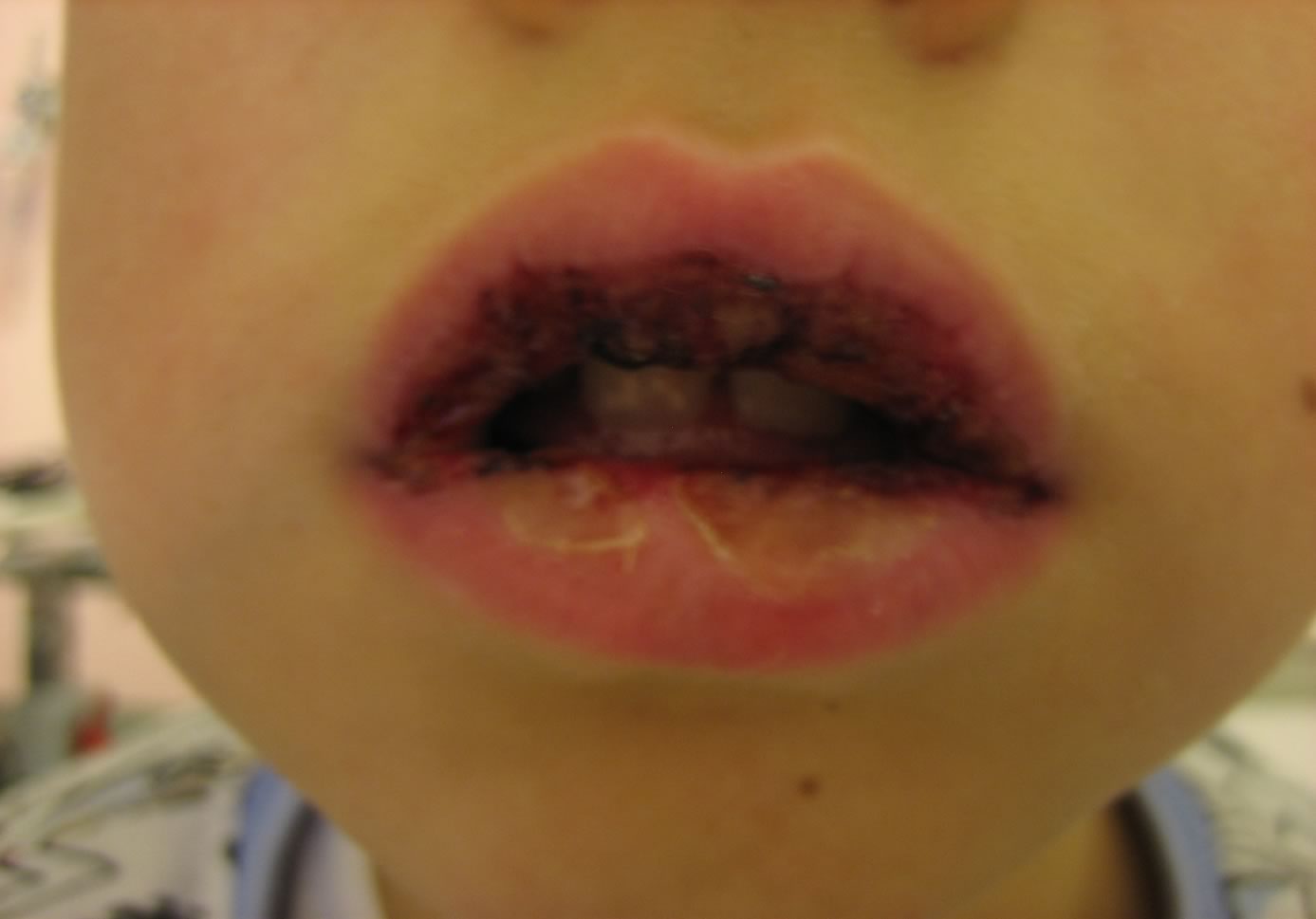 Evidente
edema delle labbra con aree di necrosi e di disepitelizzazione
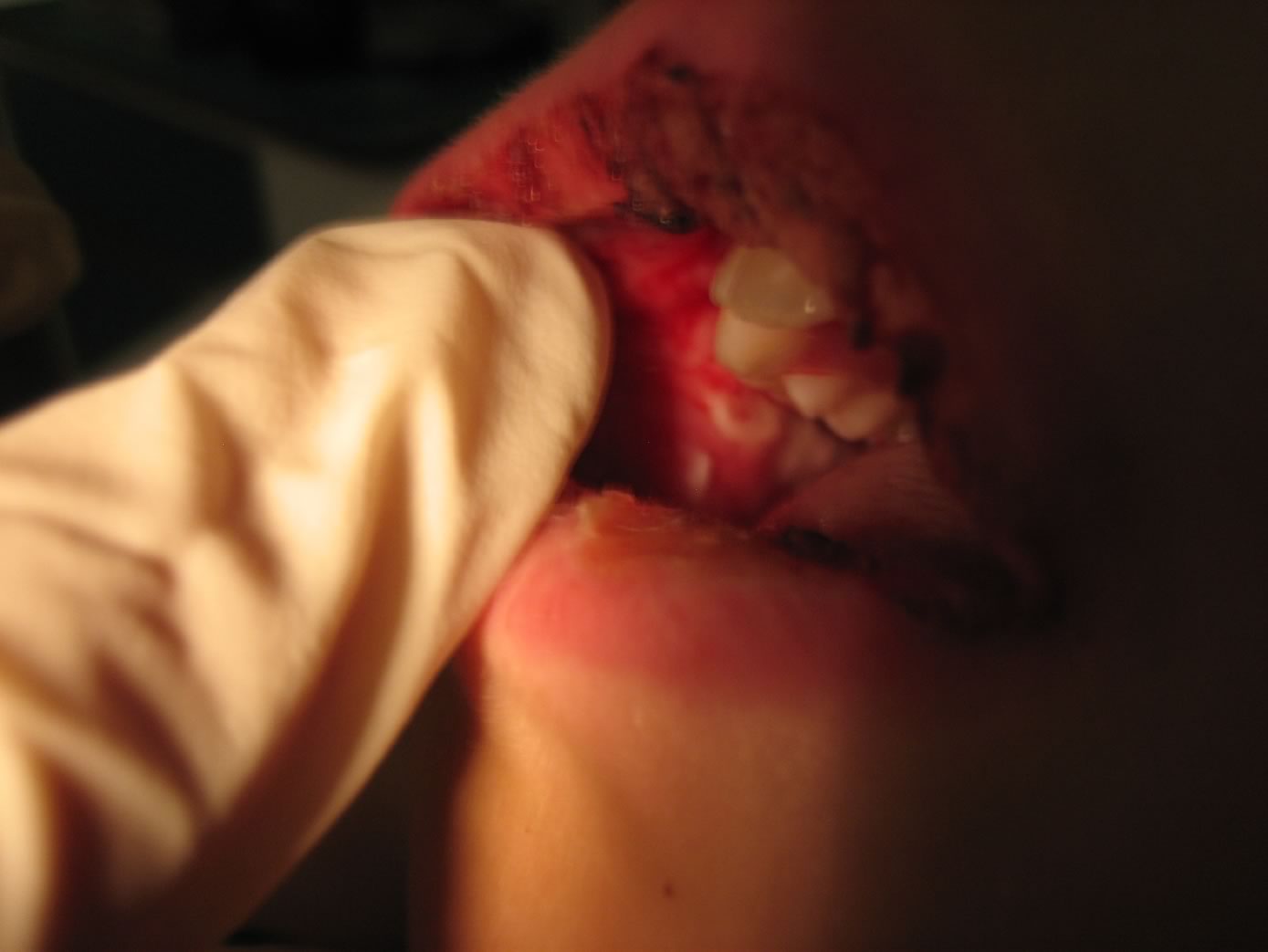 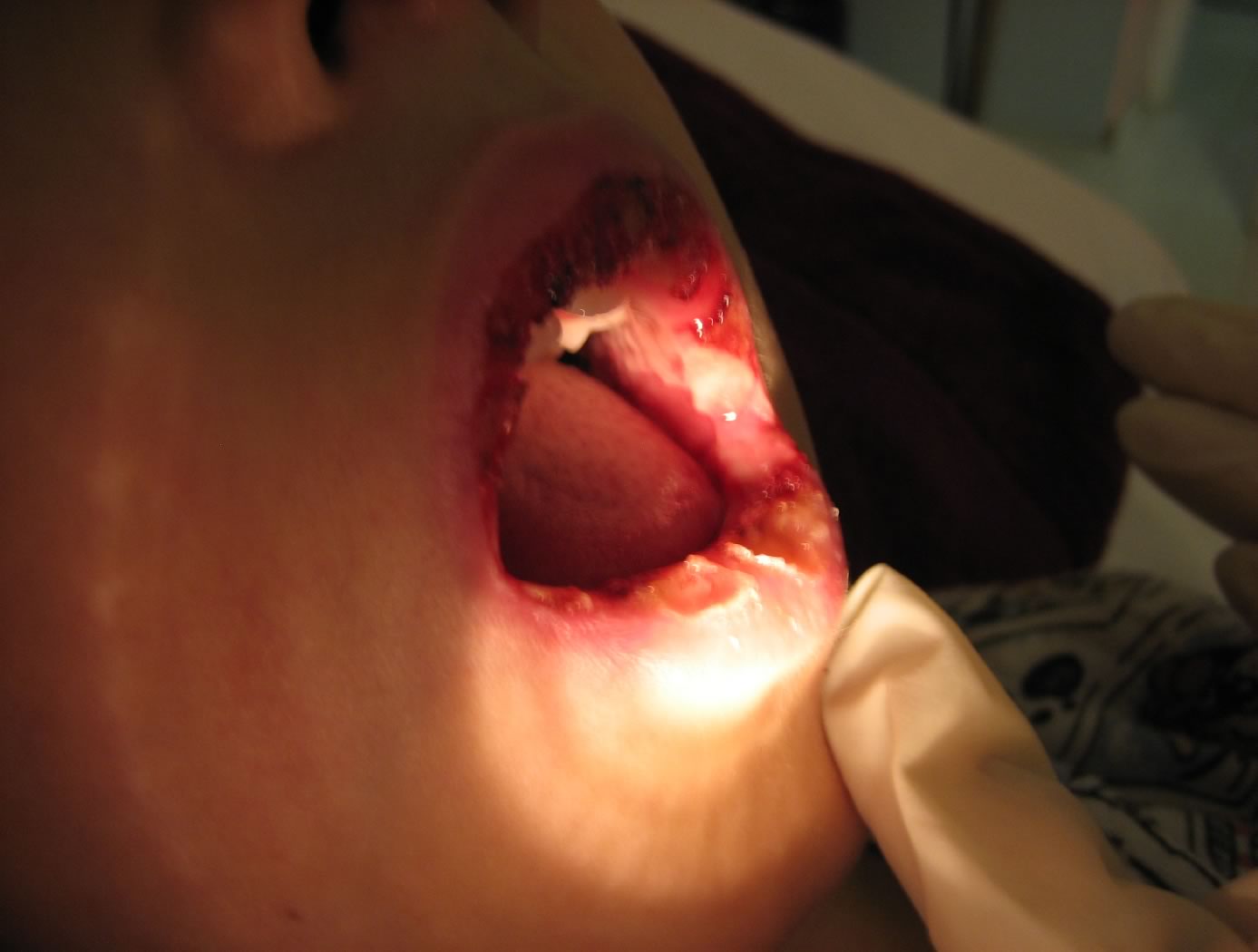 Aree
di mucosa orale con ulcerazioni, ricopertein lacune sedi da
fibrina
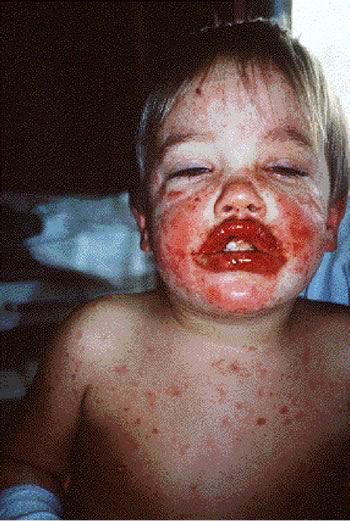  Tipico
edema delle labbra con aree di necrosi e di fibrina, associato a
lesioni cutanee diffuse caratteristicamente a tronco e volto e
che si presentano come macule eritematose e purpuriche.
 Lesioni
cutanee nella fase iniziale di vescicole che poi evolveranno in
aree necrotiche.
|
Vuoi citare questo contributo?