Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Novembre 2006 - Volume IX - numero 9
M&B Pagine Elettroniche
Pediatria per immagini
Malformazione
adenoido-cistica congenita (CCAM) polmonare
1UO
di Neonatologia e T.I.N., IRCCS Burlo Garofolo, Trieste
2UO
di Radiologia, IRCCS Burlo Garofolo, Trieste
Indirizzo
per corrispondenza: lorenzo.calligaris@inwind.it
P. è
un bambino secondogenito, nato a 39 settimane di età
gestazionale da parto spontaneo con peso adeguato (2990 grammi,
10-25° centile), a cui è stata diagnosticata attraverso
l'ecografia prenatale la presenza di una malformazione
adenoido-cistica polmonare (CCAM) coinvolgente il lobo medio e
apicale di destra, senza sbandieramento del mediastino. Alla
nascita viene trasferito in Neonatologia per la comparsa di un
moderato distress respiratorio e viene avviata una minima
supplementazione di ossigeno con pressione positiva continua
attraverso cannula naso-faringea (N-CPAP).
All'ingresso
in reparto il bambino appare in buone condizioni generali e
l'obiettività evidenzia solo una moderata polipnea, senza
altri elementi di rilievo.
|
| |
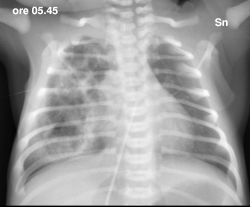
Una
radiografia del torace documenta un campo polmonare di destra in
gran parte occupato da opacità disomogenea, nel cui
contesto si evidenziano multiple areole di radio-trasparenza;
campo sinistro senza alterazioni e lieve sbandieramento del
mediastino (Figura 1). Il quadro appare
compatibile con la diagnosi prenatale di CCAM. Visto il rapido
miglioramento clinico del quadro respiratorio e la normalità
dell'emogas-analisi, la N-CPAP viene rimossa dopo poche ore. |
si
evidenziano multiple areole di radio-trasparenza; campo sx senza
alterazioni e lieve sbandieramento del mediastino. L'esame
evidenzia un catetere ombelicale che si proietta in sede atriale
(ritirato subito dopo). In sede epigastrica un area circolare di
radiotrasparenza, attribuibile a un elemento dell'incubatore. |
Per
una miglior definizione del problema, anche in vista
dell'intervento, viene effettuata una TC-polmonare che evidenzia
a destra una formazione irregolarmente ovalare di circa 5 cm di
diametro massimo, apparentemente ben marginata, con numerose
formazioni cistiche aerate circostanti (Figure
2 e Figura 3).
Alla
luce della diagnosi il bambino viene quindi sottoposto con
successo ad intervento di lobectomia parziale dei lobi medio e
superiore di destra. | 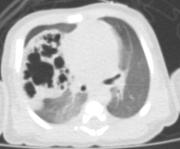 | Figure
2 a b. TC-polmonare: evidente a dx una formazione
irregolarmente ovalare di circa 5 cm di diametro massimo. | |
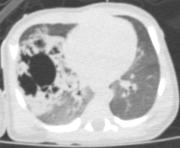 | La
formazione è apparentemente ben marginata, con numerose
formazioni cistiche aerate circostanti. |
Il
successivo decorso postoperatorio non ha presentato complicazioni
(2 giorni di ventilazione meccanica e drenaggio pleurico).
L'alimentazione già avviata con suzione autonoma prima
dell'intervento, è stata ripresa il giorno successivo
all'operazione con buona tolleranza ed il bambino è stato
dimesso, a diciotto giorni di vita, in buone condizioni generali
ed in allattamento materno esclusivo, con un programma di
follow-up presso la chirurgia del nostro istituto.
|
| |
Dall'esame
istologico giunge conferma del reperto intraoperatorio
macroscopico di CAMM di tipo I (cisti maggiore, con multiple cisti
di dimensioni limitate, rivestite da epitelio cildrico
pseudostratificato ciliato, con estesi tratti di piccole
formazioni di tipo papillare). |
Che
cosa è la CCAM
La
malformazione adenoido-cistica congenita (CCAM) è una delle
più comuni anomalie polmonari congenite. Si tratta di una rara
alterazione polmonare caratterizzata da tessuto polmonare displastico
o amartomatoso, frammisto a tessuto polmonare normale, in genere
confinato ad un solo lobo1. La lesione verosimilmente è
conseguente ad un insulto in fase embriologica, antecedente ai 50
giorni di gestazione, con alterato sviluppo delle strutture
bronchiolari terminali2.
Istologicamente
è presente poco tessuto polmonare normale e si rilevano molti
elementi ghiandolari; le cisti sono molto comuni e la cartilagine è
rara (insulto più tardivo). Le CCAM vengono classificate
sostanzialmente in 3 tipi:
- Tipo I (macrocistico): una o più grosse cisti (diametro >2 cm) delimitate da epitelio pseudostratificato ciliato; la parete della cisti contiene cellule muscolari lisce e tessuto elastico. In un terzo dei casi sono presenti cellule secernenti muco e la cartilagine è raramente presente. Buona prognosi per la sopravvivenza.
- Tipo II (microcistico): multiple piccole cisti con istologia simile al tipo I. Si associa in genere ad altre anomalie e ha una prognosi peggiore.
- Tipo III: lesione solida con strutture simil-bronchiolari delimitate da epitelio cuboide ciliato e separate da aree di epitelio cuboide non-ciliato. Forma a prognosi peggiore.
Diversamente
dalla cisti broncogena intrapolmonare, la CCAM è irrorata da
un'arteria di origine polmonare e la cisti comunica con l'albero
tracheobronchiale. L'esordio clinico è variabile; nel
neonato può manifestarsi con un distress respiratorio
progressivo severo, infezioni respiratorie e pneumotorace, nel
bambino più grande o nell'adulto possono comparire polmoniti
ricorrenti a carico di un lobo. Un massa polmonare con componente
solida prevalente si ritrova in genere nei neonati o nei prematuri e
si può associare ad idrope, ascite e polidramnios. In alcuni
casi, le cisti di piccole dimensioni rimangono asintomatiche e
rappresentano un reperto occasionale in occasione di un'indagine
strumentale.
I reperti
radiologici sono in genere diagnostici e dipendono dal tipo di
malformazione presente. La radiografia del torace può
documentare cisti singole o multiple o una massa solida. Un
pneumatocele, un enfisema lobare congenito, una cisti broncogena o
un'ernia diaframmatica entrano nella diagnosi differenziale; una
tomografia computerizzata o una risonanza magnetica, in caso di
lesione complessa, saranno d'aiuto in questo senso, anche per
valutare una malformazione evidenziata in epoca pre-natale e non
evidenziata alla radiografia standard. La valutazione istologica
rappresenta la conferma diagnostica. La diagnosi ecografia prenatale
è possibile a partire dalla 16a settimana ma, come nel nostro
caso, soprattutto dalla 21a1-3.
Laprognosi dipenderà prevalentemente dalla grandezza e
dalla progressione della lesione; la relazione tra l'aspetto
istologico e l'outcome è controversa. Lesioni di grosse
dimensioni possono determinare, attraverso la compressione del
tessuto adiacente, un'ipoplasia polmonare. In alcuni casi la
lesione può regredire o scomparire spontaneamente; in casi a
decorso più severo può invece progredire e causare
morte fetale da idrope o morte neonatale secondaria ad ipoplasia
polmonare. I reperti ecografici che suggeriscono una prognosi
peggiore includono polidramnios, idrope, ascite, sbandieramento
mediatstinico e lesioni completamente adenomatose. L'interruzione
di gravidanza viene consigliata in caso di idrope, malformazioni
severe associate ed anomalie cromosomiche. Il drenaggio di cisti di
grosse dimensioni o la rimozione di masse solide di grandi dimensioni
in utero, consente in alcuni casi un miglior sviluppo del tessuto
polmonare ed un miglioramento delle condizioni del feto2-4.
In tutti i casi di sospetta CCAM la resezione chirurgica è
raccomandata per evitare la comparsa di infezioni ricorrenti e per la
segnalazione di aumentata incidenza di neoplasie polmonari primitive;
tuttavia voci discordi, che spingono per un approccio più
conservativo in casi non sintomatici ci sono5-7. La
prognosi a lungo termine, che dipenderà dalla quota di tessuto
polmonare residuo, sarà comunque generalmente buona.
Bibliografia
- Nelson Textbook of Pediatrics. 17th edition. W.B Saunders Company, 2004:1424.
- Pathological case of the month. Congenital cystic adenomatoid malformation of the lung. Arch Pediatr Adolesc Med 2000;154(6):633-4.
- Cystic Adenomatoid Malformation of the Lung: Review of Genetics, Prenatal Diagnosis and in Utero Treatment. Am J Med Gen 2006;140A:151-55.
- Horak E, Bodner J, Gassner I, et al. Congenital Cystic Lung disease: Diagnostic and Therapeutic Considerations. Clin Pediatr 2003;42:251-61.
- Sauvat F, Michel JL, Benache A, et al. Management of Asymptomatic Neonatal Cystic Adenomatoid Malformations. J Pediatr Surg 2003;38:548-52.
- Chetcuti J, Crebbe G. CAM lungs: the conservative approch. Arch Dis Child Fetal Neonatal Ed. 2006;91;463-64.
- Jaffè A, Chitty LS. Congenital cystic malformation may not require surgical intervention. Arch Dis Child Fetal Neonatal Ed 2006;91;464.
Vuoi citare questo contributo?