
Ipertrofia
clitoridea in bambina affetta da neurofibromatosi di tipo 1
*Pediatra
di Famiglia, Milano
**Divisione
di Oncologia Pediatrica Istituto Nazionale Tumori, Milano
Indirizzo
per corrispondenza: emanuela.ballerini@crs.lombardia.it
Hypertrofy
of the clitoris in a female with neurofibromatosis 1
Key
words
neurofibromatosis
1 (NF1), hypertrophy of the clitoris, plexiform neurofibroma
Summary
We
report the case of NF1 in a 21 month old female, who presented
with hypertrophy of the clitoris. The work-up evaluations
revealed the presence of multiple pelvic and abdominal
neurofibromas and concomitant histologically assessed presacral
ganglioneuroma. Despide of multiple treatments, including
chemotherapy, radiotherapy and antiangiogenic drug, the
neurofibromas were in slow but continuous numerical and
volumetric progression. NF1 is the most frequent syndrome at
dominant autosomical transmission at variable penetrance, with a
wide spectrum of clinical presentations varying from absent or
mild symptoms, to severe disability. |
La storia
clinica di M., secondogenita, esordisce all'età di 6 mesi
quando pongo diagnosi di neurofibromatosi di tipo 1 (NF1): presenta
almeno 6 macchie caffelatte di diametro maggiore di 5 mm, comparse a
2 mesi e con graduale incremento numerico e dimensionale, ed inoltre
la madre e il fratello sono affetti dalla stessa patologia.
Durante
una visita ambulatoriale all'età di 21 mesi noto un'ipertrofia
clitoridea che onestamente ero sicura di non avere mai apprezzato
prima e che gli stessi genitori non avevano mai rilevato.
L'accrescimento
staturo-ponderale appare regolare, non sono presenti altri segni di
sviluppo puberale precoce o alterato, i meati uretrali e vaginale
sono normoconformati.
Stupita e
perplessa per il riscontro clinico (attivazione dell'asse
ipotalamo-ipofisario secondaria a glioma delle vie ottiche, possibile
complicanza della NF1? Patologia cromosomica?), invio la bambina per
una visita endocrinologica, a cui fa seguito, a distanza di alcuni
mesi, per motivi organizzativi, un Day
Hospital: i test di funzionalità endocrina risultano
nella norma, vi è una stazionarietà del quadro clinico,
un'ecografia pelvica non permette di riconoscere utero e ovaie e
documenta una formazione parenchimatosa a forma di clessidra, non ben
interpretabile, tra la vescica e il retto mentre un'analisi del
cariotipo risulta normale per il sesso femminile.
Un
successivo controllo ecografico , programmato dai Colleghi
Ospedalieri, evidenzia una tumefazione pelvica a profili bozzuti,
all'estremità craniale di quella che sembra parte del fondo
uterino, che presenta dimensioni e morfologia conservate.
Contemporaneamente compare prurito a livello della coscia di sinistra
e visitando M. ora è presente edema vulvare, aumento
dimensionale del gluteo di sinistra e area di infiltrazione cutanea a
livello perianale.
Nel
sospetto di una neoformazione tumorale condizionante una
compressione delle strutture a livello pelvico (sarcoma associato
a NF1?) viene ricoverata all'età di 2 anni e 9 mesi presso
la Divisione di Oncologia Pediatrica dell'Istituto Nazionale
Tumori di Milano.
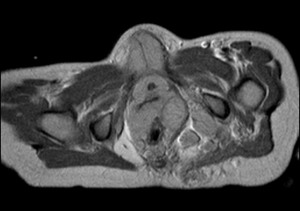
Gli
esami ematochimici di routine sono normali, alcuni indici tumorali
(alfa-feto-proteina, beta-HCG, NSE) risultano negativi, mentre la
RMN dell'addome, oltre a tessuto endopelvico che circonda l'utero,
l'uretra e le pareti del retto, documenta noduli multipli, da
pochi mm a 3 cm, all'emergenza e lungo il decorso di plessi e
radici nervose sacrali, soprattutto a sinistra, nodulazioni nella
muscolatura glutea e nel muscolo otturatore interno di sinistra e
piccoli noduli plurimi lungo il decorso del nervo sciatico di
sinistra (Figura, vedi a lato). |
La bimba
viene sottoposta a laparotomia trasversa sovrapubica: è
confermata macroscopicamente la presenza delle nodulazioni multiple
che si estendono dalla pelvi a tutto il peritoneo, compatibili con
neurofibromi plessiformi, che non vengono biopsiate perché il
trauma potrebbe esercitare un effetto stimolatore sulla crescita
degli stessi mentre la biopsia escissionale di una lesione
presacrale, che ha un differente aspetto macroscopico, permette di
porre una diagnosi istologica di ganglioneuroma, cioè di un
tumore benigno che interessa le strutture nervose del parasimpatico.
La storia
naturale dei neurofibromi plessiformi non è ancora ben
conosciuta, possono rimanere silenti per molti anni e l'unica valida
possibilità terapeutica in caso di importanti disturbi
funzionali è considerata la totale escissione chirurgica,
indicata anche per il ganglioneuroma, quasi sempre però
impossibile, come anche in questo caso, sia per la numerosità
e l'estensione delle lesioni sia per le probabili numerose proiezioni
digitiformi all'interno dei tessuti normali circostanti. M .viene
dimessa con l'indicazione di periodici controlli nel tempo. Dall'età
di 3 anni e 4 mesi si sono manifestati ricorrenti momenti di
progressione clinica delle lesioni, condizionanti disturbi funzionali
ed estetici.
Data la
sede e l'estensione dei neurofibromi, sempre esclusa la possibilità
di procedere a un intervento chirurgico, sono stati da allora
effettuati diversi tentativi terapeutici allo scopo di bloccarne
l'evoluzione, con temporanee stabilizzazioni di malattia: terapia
metabolica con MIBG-I-131, chemioterapia con cisplatino ed etoposide,
e poi ciclofosfamide e chetotifene, vinorelbina e methotrexate. A sei
anni di età, per l'ulteriore progressione dimensionale dei
neurofibromi e con un'obiettività caratterizzata da una massa
al gluteo di sinistra, importante edema vulvare e una massa
addominale e pelvica di 10x12cm condizionante iniziale idronefrosi, è
stato intrapreso un nuovo tentativo terapeutico con talidomide 75
mg/die per os, farmaco inibente sia il Tumor Necrosis Factor
sia l'angiogenesi, con, a oggi, una sostanziale stazionarietà
della malattia. Il follow-up oncologico è invece negativo per
segni di progressione del ganglioneuroma o per la comparsa di
neoplasie maligne associate alla NF1.
In tutti
questi anni la qualità di vita di M. può essere
definita buona. I periodi di ricovero e di trattamento ambulatoriale
sono stati brevi e con modesta tossicità, ha frequentato
regolarmente la scuola materna, pratica attività sportiva e la
famiglia, coinvolta attivamente nelle decisioni terapeutiche, ha
sempre manifestato un valido appoggio ai medici che si stanno
prendendo cura della piccola.
Discussione
La NF1,
termine che oggi sostituisce il vecchio nome di malattia di Von
Recklinghausen, è la più frequente sindrome a
trasmissione autosomica dominante con una incidenza di 1 su
2.500-3.000 nati ed una prevalenza di circa 1 su 4.000-5.000
individui nella popolazione generale1; nel 50% è dovuta a
mutazione de novo.
La
diagnosi viene posta quando sono soddisfatti due o più dei
criteri clinici proposti dal National Institute of Health nella
Consensus Conference di Bethesda del 19872 (Tabella)
e le attuali indicazioni, non però condivise da tutti gli
autori, sottolineano che non è consigliabile l'impiego di
esami strumentali alla diagnosi o per il monitoraggio di complicanze
non evidenti all'esame clinico o in assenza di un minimo sospetto
diagnostico1,3.
6
o più macchie caffelatte con diametro > 5 mm nel periodo
prepubere o 15 mm nel periodo postpubere |
2
o più neurofibromi di ogni tipo o un neurofibroma
plessiforme |
lentiggini
ascellari o inguinali |
2
o più noduli di Lisch ( amartomi dell'iride) |
una
specifica lesione ossea come la displasia dello sfenoide o
l'assottigliamento della corticale delle ossa lunghe con o senza
pseudoartrosi |
un
parente di primo grado affetto da NF1 ( genitore, fratello,
figlio) |
Devono
essere presenti due o più criteri |
Si
distinguono segni clinici maggiori, presenti nella maggioranza degli
individui affetti e che fanno parte dei criteri diagnostici (macchie
caffelatte, lentiggini ascellari o inguinali, noduli iridei di Lisch
e neurofibromi cutanei o sottocutanei); segni clinici minori,
presenti in una discreta percentuale degli affetti (macrocefalia,
bassa statura, anomalie toraciche tipo pectus excavatum o carenatum)
e complicanze, poco frequenti ma spesso gravi e invalidanti (disturbi
cognitivi e dell'apprendimento, neurofibromi plessiformi, scoliosi,
pseudoartrosi, malformazioni cardio-vascolari e ipertensione
arteriosa, glioma delle vie ottiche4, tumori del sistema nervoso
centrale, neurofibrosarcomi).
La
penetranza della malattia è elevata, arriva virtualmente al
100% attorno ai 5-6 anni di vita, età in cui tutti i soggetti
affetti presentano i segni clinici della malattia mentre
l'espressività è molto variabile, da casi con lieve
sintomatologia a casi con complicanze molto gravi (10%).
Tra le
complicanze non possiamo dimenticare la patologia neoplastica5:
i soggetti affetti da NF1 hanno una frequenza relativa alta per
glioma delle vie ottiche, neurofibrosarcoma e feocromocitoma, con
rischio assoluto basso, mentre la patologia neoplastica propria
dell'età infantile e di quella adulta non si presenta con
frequenza maggiore. Una spiegazione potrebbe essere legata ad una
mutazione a carico del gene localizzato in sede pericentrometrica del
braccio lungo del cromosoma 17, posizione 17q11.2, che codifica una
proteina, la neurofibromina (2.818 aa), ben rilevabile nei
microtubuli citoplasmatici delle cellule nervose e che è
regolatrice della crescita e della differenziazione cellulare, con
funzione di tumor-suppressor. Una mutazione con codifica di una
neurofibromina meno efficace potrebbe essere causa di proliferazione
cellulare incontrollata con predisposizione a sviluppare malignità
per diminuzione dell'effetto di controllo negativo sulla crescita
cellulare6.
I
neurofibromi sono tumori benigni che possono interessare qualunque
nervo, formati da cellule di Schwann, fibroblasti, mastociti, cellule
endoteliali; quando contengono un'abbondante matrice extracellulare e
posseggono interdigitazione verso le strutture adiacenti sono
denominati neurofibromi plessiformi. I neurofibromi plessiformi in
genere esordiscono nei primi anni di vita ma a volte rimangono
silenti per lungo tempo e diventano clinicamente evidenti più
tardi7. Le lesioni a comparsa in età infantile più
facilmente coinvolgono grossi tronchi nervosi e vasi venosi,
arteriosi e linfatici, oppure dislocano organi interni condizionando
stasi con edema e ipertrofia dei tessuti, come nel caso di M., con
coinvolgimento per esempio vulvare e gluteo.
In caso
di progressione delle lesioni condizionanti disturbi funzionali od
estetici severi vengono effettuati tentativi radioterapici o
chemioterapici o con farmaci antiinfiammatori, che sinora non si sono
dimostrati sicuramente efficaci8,9. All'incirca nel 5% dei
casi, più frequentemente quando un neurofibroma è
presente da lungo tempo o è assai esteso, possono trasformarsi
in lesioni maligne, cioè in neurofibrosarcomi10.
Rivalutando
la storia clinica di M. non possiamo non rilevare due criticità:
tra il riscontro clinico ambulatorialmente di ipertrofia clitoridea,
confermata da consulenza endocrinologica, e l'esecuzione di indagini
mirate trascorrono, per motivi organizzativi, alcuni mesi ed il
sospetto di una patologia oncologica, che è segnalata come una
possibile complicanza della NF1, emerge non dalla
prima
ecografia pelvica, che orienta verso una patologia cromosomica , ma
dalla seconda indagine radiologica. Questa difficoltà
diagnostica, data la storia naturale dei neurofibromi plessiformi,
non ha inficiato le successive decisioni ma un messaggio importante
che deve emerge da questo caso clinico è che, data l'estrema
variabilità del decorso e della gravità della NF1, con
possibilità di complicanze molto gravi, in presenza di una
qualunque alterazione dell'esame clinico è auspicabile sempre
per prima cosa escludere una patologia oncologica.
Bibliografia
1.
Ruggieri M, Tenconi R. Le neurofibromatosi. Associazione
Neurofibromatosi. Grafiche Step 2003:1- 95.
2.
National Institutes of Health Consensus Development Conference
Statement.Neurofibromatosis. Arch Neurol 1988;45:575-78.
3.
Committee on Genetics. Health supervision for children with
neurofibromatosis. Pediatrics 1995(2);96:368-72.
4. Virdis
R, Street ME, Tripodi C. Growth and pubertal disorders in
neurofibromatosis type 1. J Pediatr Endocrinol Metab 2003;16
(2):289-92.
5. Korf
B. Malignancy in Neurofibromatosis Type 1. The Oncologist
2000;5:477-85.
6. Zhu
Y,Parada LF. Neurofibromin, a tumor suppressor in the nervous system.
Exp Cell Res 2001;264:19-28.
7.
Tonsgard J, Kwak S, Short P, Dachman AH. CT imaging in adults with
neurofibromatosis-1: frequent asymptomatic plexiform lesions.
Neurology 1988(50);1755-60.
8. Packer
RJ, Gutmann DH, Rubenstein A, et al. Plexiform neurofibromas in NF1:
toward biologic-based therapy. Neurology 2002;58:1461-70.
9. Gupta
A, Cohen BH, Ruggieri P, Packer RJ, Phillips PC. Phase 1 study of
thalidomide for the treatment of plexiform neurofibroma in
neurofibromatosis 1. Neurology 2003; 60:130-32.
10.
Neville H.L, Seymour-Dempsey K, Slopis J, et al. The role of surgery
in children with neurofibromatosis. Journal of Pediatric Surgery
2001;36:25-9.
Vuoi citare questo contributo?
E. Ballerini, R. Luksch. IPERTROFIA CLITORIDEA IN BAMBINA AFFETTA DA NEUROFIBROMATOSI DI TIPO 1.
Medico e Bambino pagine elettroniche 2006; 9(9)
https://www.medicoebambino.com/_neurofibromi_neurofibromatosi_clinico_patologia_plessiformi_diagnosi_lesioni_complicanze_malattia