Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Ottobre 2007 - Volume X - numero 8
M&B Pagine Elettroniche
Pediatria per l'ospedale
Il
Neuroblastoma (parte seconda)
Membro
della Commissione Nazionale Vaccini
Indirizzo
per corrispondenza: bartolozzi@unifi.it
Il
neuroblastoma rappresenta ancor oggi uno dei tumori più
misteriosi: ne sono prova l'estrema eterogeneicità del
quadro clinico e il ventaglio della prognosi che va dalla relativa
benignità dello stadio 4S all'estrema gravità dello
stadio 4, che comporta tutt'oggi una letalità del 40%
nonostante l'uso di farmaci potenti. L'interesse dei ricercatori
è documentato dal numero di pubblicazioni sull'argomento
nel corso degli anni: alla voce NEUROBLASTOMA su PubMed nell'arco
di 55 anni ho trovato 24.757 studi, con un massimo di 1.300 nel
2006.Una recentissima revisione, comparsa sul fascicolo del 23
giugno 2007 del Lancet, offre la possibilità di rivedere i
vari aspetti di questo tumore (Maris JM, Hogarthy MD, Bagatell R,
Cohn SL. Neuroblastoma. Lancet 2007, 369:2106-20).
Principi
di terapia d'attacco
I metodi
di trattamento del neuroblastoma includono:
- La chirurgia
- La chemioterapia
- La radioterapia
- La terapia biologica
- La semplice osservazione, in casi accuratamente selezionati.
Il
sistema di stratificazione del rischio del Children's Oncology
Group comprende pazienti, suddivisi a seconda dell'età,
dello stadio INSS (International Neuroblastoma Staging System),
dell'istopatologia del tumore, dell'indice DNA e dello stato del
gene MYCN, in base ai quali i pazienti vengono assegnati a uno dei
tre gruppi:
- Basso rischio
- Rischio intermedio
- Alto rischio
A seconda
dello stadio, viene impiegato un trattamento più o meno
intenso. Come si vede nella Figura 4, gli schemi usati negli ultimi
15 anni sono risultati molto utili per l'identificazione di gruppi
di pazienti con prognosi molto differenti.
Figura
4. Sopravvivenza di pazienti con neuroblastoma, suddivisi per i
gruppi a rischio. Pazienti trattati dal 1986 al 2001 nel Children's
Cancer Group, Pediatric Oncology Group e Children's Oncology Group,
che furono classificati in basso, intermedio e alto rischio, a
seconda dei dati clinici e biologici.
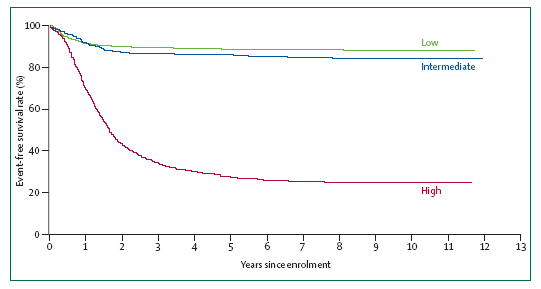
Quindi,
semplificando, si devono considerare due importanti parametri:
- Innanzitutto, si deve essere stabilire se rimane un tumore residuo dopo la resezione chirurgica. In questo caso, l'integrazione fra i dati clinici e biologici è fondamentale per facilitare l'identificazione di come si comporterà il tumore residuo. In molte occasioni è risultato che i parametri biologici sembrano essere più importanti degli aspetti clinici caratteristici, come predittori della prognosi. Va ricordato inoltre che il neuroblastoma ha una forte tendenza a regredire o a differenziarsi.
- Al contrario, alcuni tumori localizzati possono essere resecati quasi completamente. Ma anche in questo caso aspetti biologici poco favorevoli confermano la necessità di una terapia intensiva adiuvante.
Tumori
locoregionali
Generalmente
i neuroblastomi localizzati presentano parametri biologici
favorevoli, per cui la maggior parte di essi viene trattata con
successo con la sola chirurgia. Vi sono prove che una parte dei
tumori localizzati regredisce spontaneamente, per cui questi pazienti
possono essere tenuti sotto osservazione senza alcun trattamento.
Riprese locali possono essere ugualmente trattate chirurgicamente.
Le
ricadute metastatiche sono rare e spesso sono affrontate con successo
con la chemioterapia.
Il
trattamento di pazienti con forme localizzate, ma con aspetti
biologici sfavorevoli, specialmente di amplificazione del MYCN,
rimane controverso. Sebbene questi bambini abbiano una prognosi
sostanzialmente cattiva in confronto ai pazienti con malattia
localizzate e mancanza di amplificazione del MYCN, una parte di essi
presenta una remissione di lunga durata dopo la sola chirurgia.
Questi rari casi tuttavia richiedono una continua sorveglianza, per
chiarire quale sia il migliore trattamento.
La cura
dei tumori locoregionali più invasivi (INSS stadio 3) rimane
controversa. Classicamente questi pazienti ricevono una chemioterapia
moderatamente intensiva con lo scopo di facilitare la successiva
rimozione chirurgica. I tumori con invasione locoregionale e con
sfavorevoli aspetti biologici, richiedono una terapia intensiva
multimodale.
Tumori
metastatici
Quasi la
metà di tutti i pazienti con neuroblastoma hanno già
una disseminazione della malattia, al momento della diagnosi.
La
maggior parte dei pazienti con lo stadio 4S (lattanti con
iperploidia, istologia favorevole, con una sola copia di MYCN)
rientra nella categoria a basso rischio con una sopravvivenza che va
dall'85 al 95%. Tuttavia, i pazienti diagnosticati nei primi due
mesi di vita sembrano particolarmente vulnerabili a una
compromissione respiratoria, secondaria all'epatomegalia
rapidamente progressiva. Inoltre una parte di pazienti in stadio 4S
hanno aspetti biologici non favorevoli, come un'amplificazione
MYCN: essi hanno spesso una progressione rapida del tumore o
eventuali ricadute di malattia, simili a quelle che si vedono nello
stadio 4. Queste differenze sottolineano, una volta di più,
l'importanza della valutazione biologica del tumore in pazienti in
stadio 4S di malattia.
Il
trattamento di bambini di maggiore età con neuroblastoma
altamente disseminato (stadio 4), rimane una delle maggiori sfide per
gli oncologi pediatri. Comunemente vengono usati diversi agenti, che
includono il cisplatino, l'etoposide, la doxorubicina, la
ciclofosfamide e la vincristina. Da più di 10 anni si utilizza
la combinazione di topotecan e ciclofosfamide nelle ricadute.
La
successiva fase di consolidamento della terapia ha lo scopo di
eliminare ogni rimanente parte del tumore, usualmente con agenti
citotossici mieloablativi, con successivo salvataggio mediante
cellule staminali.
Poiché
spesso/di frequente si verifica una ricaduta dopo il trapianto
autologo, la terapia biologica è stata aggiunta ai correnti
regimi di trattamento per trattare la malattia residua minima
persistente. Molti nuovi agenti, rivolti unicamente al trattamento
del neuroblastoma, possono essere efficaci per eliminare la malattia
residua minima. È noto che i retinoidi sono una classe di
composti che inducono la differenziazione terminale delle cellule di
neuroblastoma in vitro.
Valutazione
della risposta al trattamento
Al
termine del trattamento, le catecolamine urinarie e gli studi con
immagini (TC, RM, esame del midollo, della corticale ossea o di
ambedue) vanno usati per monitorare la ricomparsa del tumore.
Predisposizione
al neuroblastoma
Una
storia familiare di neuroblastoma si raccoglie nell'1-2% dei casi.
In questi rari casi si suppone sia in gioco un'eredità
autosomico dominante con penetranza incompleta. Studi tradizionali
hanno identificato il braccio corto del cromosoma 16, come locus
predisponente, anche se non è stato identificato un singolo
gene. Il neuroblastoma è stato visto anche in molti
riarrangiamenti costituzionali, incluse le delezioni. Ne deriva che
la predisposizione al neuroblastoma è geneticamente
eterogenea, per cui, per l'inizio della tumore-genesi, possono
essere necessarie alterazioni multiple.
I
recettori della neurotrofina (NTRK1, NTRK2 e NTRK3 codificanti TrkA,
TrkB e TrkC) e i loro ligandi (NGF, BDNF e neurotrofina-3
rispettivamente) sono importanti regolatori della sopravvivenza,
della crescita e delle differenziazione delle cellule nervose. La
loro espressione temporo-spaziale interessa lo sviluppo del sistema
simpatico e si correla con il fenotipo del neuroblastoma.
Nuove
strade per la malattia in ricaduta
Sebbene
vi siano terapie, altamente efficaci per pazienti con malattia a
basso rischio o a rischio intermedio con ricadute locali, malattie
ricorrenti in pazienti con neuroblastoma ad alto rischio rimangono
una sfida per l'oncologo pediatra. Durante gli ultimi anni tuttavia
si è sviluppata un'ampia scelta di nuovi agenti o di
combinazioni da usare in pazienti con ricadute ad alto rischio.
Agenti
citotossici
Gli
inibitori della topoisomerasi 1, tipotecam e irinotecan, sono usasti
spesso precocemente nelle ricadute, per la loro efficacia e per il
loro accettabile profilo di tossicità. L'efficacia del
topotecam aumenta quando viene utilizzato insieme a basse dosi di
ciclofosfamide.
Un nuovo
agente citotossico, l'ABT-751, somministrabile per bocca, si è
dimostrato utile nel neuroblastoma refrattario alle altre cure.
Radionuclidi
Poiché
i neuroblastomi in ricaduta sono spesso sensibili alle radiazioni, si
è sviluppato un forte interesse riguardo l'uso delle
molecole radioattive, che selettivamente si concentrano nelle cellule
del neuroblastoma. Per questa ragione sono stati attaccati all'MBIG
dei radionuclidi. Basse dosi di I131-MBIG si sono dimostrate utili
nei trattamenti palliativi.
Immunoterapia
GD2 è
un disialogangloside espresso nella maggioranza dei neuroblastomi. In
terapia sono stati usati vari anticorpi monoclonali, diretti verso
questa sostanza. Altre strategie immunologiche includono strategie
con DNA vaccinazioni, con vaccinazioni anti-idiotipo e linfociti
citolitici.
Retinoidi
Al
momento attuale l'obiettivo primario è quello d'identificare
il composto retinoide che dà i migliori risultati senza
elevata tossicità. Il Fenretinide è quello che uccide
il maggior numero di cellule neuroblastomatose, anche se resistenti
agli altri retinoidi. Si sta valutando la possibilità di
somministrare il farmaco per bocca.
Inibitori
dell'angiogenesi.
La
vascolarità del tumore si correla con un fenotipo aggressivo
di neuroblastoma, tanto che oggi gli inibitori dell'angiogenesi
rappresentano un'opzione attraente.
Inibitori
della tirosina chimasi
Il
CEP-701, una piccola molecola, inibitrice della titosin-chinasi, ha
dimostrato un evidente effetto inibitorio sulla crescita del
neuroblastoma.
Vuoi citare questo contributo?