Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Novembre 2006 - Volume IX - numero 9
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Diagnosi
di malattia di Rendu-Osler-Weber in bambina con cianosi e
teleangiectasie cutanee
Divisione
di Ematologia, Dipartimento di Scienze Pediatriche e
dell'Adolescenza, Università degli Studi di Torino,
Ospedale
Infantile Regina Margherita
Indirizzo
per corrispondenza: paola.quarello@unito.it
Diagnosis
of Rendu-Osler-Weber Syndrome in a child with cyanosis and
muco-cutaneous telangiectasias
Key
words
Hereditary
Hemorrhagic Telangiectasia, Cyanosis, Arteriovenous malformations
Summary
We
report the case of an 9 years old child with cyanosis and
clubbing. Her mother was affected by Hereditary Hemorrhagic
Telangiectasia (HHT) with muco-cutaneous telangiectasias and
epistaxis. Decreased level of oxygen in arterial blood was
detected and CT scan showed a pulmonary arteriovenous
malformation (AVM).
The
young patient underwent transcatheter embolization with good
results. Diagnosis of HHT was done on the basis of HHT diagnostic
criteria. Clinical features, diagnostic approaches and
therapeutic strategies during the pediatric age are discussed. |
F. è
una bambina di 9 anni la cui mamma si rivolge al pediatra di famiglia
perché ha notato la presenza di una deformazione alle dita
delle mani. La bambina, in precedenza, ha sempre goduto di buona
salute e ha raramente necessitato di una visita pediatrica.
L'anamnesi fisiologica risulta nella norma: F. è nata a
termine per parto eutocico da gravidanza decorsa fisiologicamente. Le
condizioni alla nascita erano buone, il peso neonatale adeguato
all'età gestazionale (3250g). Ha eseguito le vaccinazioni
previste dalla legge. Non viene effettuata un'approfondita anamnesi
familiare. Alla visita il pediatra di famiglia constata buone
condizioni generali ma identifica una deformazione alle dita delle
mani segnalata come dita a bacchetta di tamburo; riscontra
inoltre una evidente cianosi labiale e periungueale. All'esame
obiettivo non sono presenti altre anomalie. Il pediatra, in prima
ipotesi, sospetta una patologia cardiaca; la bambina effettua
pertanto una visita specialistica con l'esecuzione di
elettrocardiogramma ed ecocardiogramma che evidenziano unicamente la
presenza di uno sdoppiamento fisiologico del secondo tono. La
pressione arteriosa risulta normale; non viene rilevata la
saturazione dell'ossigeno.
Esclusa
la presenza di una patologia cardiaca, il pediatra di famiglia,
prescrive esami ematochimici con riscontro di eritrocitosi (Hb=16,3
g/dl; Hct=46,2 fl). In considerazione della policitemia associata a
cianosi, F. viene inviata per consulenza ematologica presso la nostra
Divisione. Approfondendo l'anamnesi la mamma riferisce di
manifestare frequenti epistassi che costituiscono, insieme alla
presenza di numerose teleangectasie cutanee e mucose, l'unica
manifestazione clinica della malattia di Rendu-Osler-Weber, ereditato dal
padre. Segnala, inoltre, che la bambina ha sempre presentato, fin
dalla prima infanzia, lieve cianosi labiale e periungueale associata
a facile affaticamento durante lo sforzo.
F.
mostra all'esame obiettivo buone condizioni generali, cianosi
periorale e alle estremità; lingua e mucosa orale
lievemente cianotiche. Dita a bacchetta di tamburo. Piccola
teleangectasia al naso, numerosi spider naevi al volto, mani e
avambracci, vermiglio labiale; fini teleangectasie non a chiazze
alle regioni zigomatiche. All'apparato cardiaco: toni validi,
ritmici, lieve impurità sistolica. All'apparato
respiratorio murmure vescicolare presente su tutto l'ambito
polmonare. Addome trattabile, non dolente, organi ipocondriaci in
limiti. Pressione arteriosa in range (100/75 mmHg). La saturazione
dell'ossigeno in aria ambiente risulta, invece, gravemente
ridotta (86-87%) sia in clinostatismo che in ortostatismo. Il
quadro clinico, la policitemia, la ridotta saturazione
dell'ossigeno considerati alla luce del dato anamnestico della
malattia di Rendu-Osler-Weber nella mamma, ci hanno indotti a
sospettare la presenza di una malformazione vascolare polmonare,
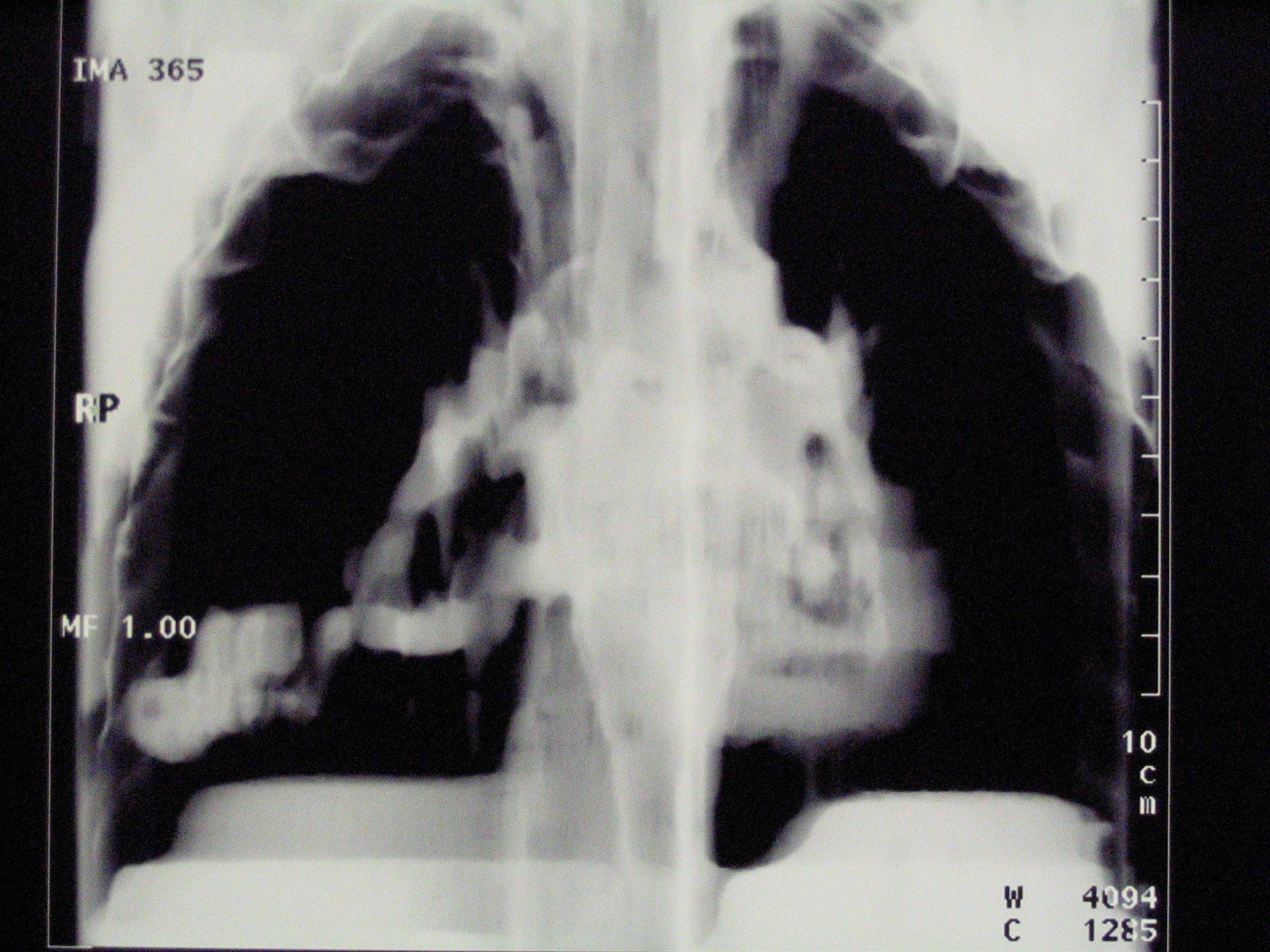
compatibile con la malattia. Abbiamo pertanto effettuato una
tomografia computerizzata del torace (TC) che ha evidenziato, a
carico del parenchima polmonare di destra, la presenza di una
grossolana anomalia vascolare a prevalenza venosa (vedi Figura
a lato). |
Per
escludere la presenza di analoghe malformazioni vascolari in altri
distretti, sono state effettuate inoltre una TC addominale e
cerebrale, risultate entrambe nella norma. La bambina è stata
sottoposta a embolizzazione percutanea della malformazione polmonare
artero-venosa (MAV) con esito positivo. Nel post-operatorio si è
assistito a graduale scomparsa della cianosi e della astenia
sotto-sforzo. In base alla caratteristiche clinico-anamnesitche è
stata posta diagnosi di malattia di Rendu-Osler-Weber. È
attualmente in corso l'indagine molecolare.
La
malattia di Rendu-Osler-Weber
La
malattia di Rendu-Osler-Weber o Teleangectasia Emorragica Ereditaria
(HHT, #187300) è una patologia a trasmissione autosomica
dominante caratterizzata da una displasia vascolare sistemica. La
prevalenza è di 1-2 individui su 10.000, con uguale incidenza
nei due sessi1. Le manifestazioni cliniche caratteristiche
sono lesioni angiodisplastiche mucocutanee o viscerali
(teleangectasie e malformazioni artero-venose), variamente
distribuite a livello di tutto il sistema cardiovascolare. Dal punto
di vista istogenetico le lesioni patologiche sono caratterizzate da
vasi dilatati, verosimilmente venule post-capillari, la cui parete è
costituita da uno strato endoteliale adeso a una membrana basale
continua2. HHT è stata descritta per la prima volta
nel diciannovesimo secolo come un disordine caratterizzato da
epistassi e sanguinamenti gastrointestinali associati ad anomalie
vascolari3-5. Nel 1896 il medico francese Rendu la
identificò come malattia ereditaria distinta dall'emofilia6.
Nel ventesimo secolo Osler e Weber definirono il quadro completo
delle sue manifestazioni cliniche7,8.
Basi
molecolari
HHT è
una patologia a trasmissione autosomica dominante con una penetranza
incompleta e un'espressività variabile. Recentemente sono
stati identificati due geni causali, endoglina e Activin Like Kinase
1 (ALK1), codificanti entrambi per recettori TGF beta,
rispettivamente di tipo III e di tipo I, espressi esclusivamente su
cellule dell'endotelio vascolare2,9. Mutazioni in tali
geni permettono di distinguere la patologia in due sottotipi HHT1 e
HHT2.
Clinica
e criteri diagnostici
Le
lesioni vascolari si esprimono clinicamente come teleangectasie e
malformazioni artero-venose di cute, mucose e visceri (polmoni,
fegato ed encefalo). I criteri clinici diagnostici per la malattia di
Rendu Osler Weber sono stati elaborati nel 2000 da un gruppo di
studio internazionale10 e sono riportati in Tabella.
La difficoltà di confermare il sospetto diagnostico in base a
soli criteri clinici in età pediatrica, risulta evidente se si
considera che le manifestazioni non sono in genere presenti alla
nascita e compaiono progressivamente all'aumentare dell'età.
È stato riportato che all'età di 16 anni il 29% dei
pazienti non presenta alcun segno clinico suggestivo di HHT; all'età
di 40 anni oltre il 90% dei soggetti affetti è clinicamente
sintomatico1,11.
1.
Epistassi spontanee e ricorrenti |
2.
Teleangiectasie multiple presenti nelle sedi tipiche della
malattia
(labbra,
cavo orale, naso e polpastrelli delle dita) |
3.
Fistole, MAV o altre anomalie vascolari agli organi interni
(stomaco,
intestino, fegato, cervello, midollo spinale o polmoni) |
4.
Familiarità positiva (un parente di 1° grado con HHT
definita) |
Diagnosi:definita, ≥3 su 4
criteri; sospetta, 2 su 4 criteri; improbabile, <2
su 4 criteri |
Tabella.
Criteri diagnostici dell'HHT |
Epistassi
Le
epistassi sono un disturbo molto frequente in età pediatrica.
La diagnosi differenziale tra sanguinamento nasale da HHT ed
epistassi benigna del bambino può essere effettuata con una
semplice visita ORL; le teleangectasie della mucosa nasale sono,
infatti, facilmente distinguibili dalle varici del Locus Valsalvae
comunemente evidenziate in bambini con epistassi frequenti.
Rappresentano
il segno clinico più precoce e frequente della malattia
(riportato nel 78-96% dei soggetti adulti). Sono state riportate in
circa il 50% dei soggetti pediatrici HHT12. Presentano
comunemente tendenza all'aggravamento con il progredire dell'età13.
Il
sintomo ha notevole variabilità di espressione clinica
individuale e intrafamiliare, sia in termini di frequenza (da uno-due
episodi/anno a numerosi episodi giornalieri) che di intensità
e durata. Il trattamento di supporto può pertanto variare
dalla sola supplementazione marziale alla necessità di
emotrasfusione. Non sono al momento disponibili dati di letteratura
riguardo all'utilizzo e all'efficacia nel bambino dei diversi
trattamenti comunemente impiegati in età adulta14-17.
Recente la segnalazione di buoni risultati ottenuti su 4 pazienti
pediatrici con laser terapia12.
Teleangiectasie
cutanee e mucose
Le
teleangectasie cutanee e mucose sono piccole lesioni rilevate, assai
fragili, che scompaiono alla digitopressione, di colore rosso
violaceo, con un diametro di 1-3 mm, prevalentemente localizzate a
livello di guance, labbra, lingua, palato, palpebre, congiuntive,
polpastrelli e letto ungueale. Riportate in circa il 75% dei pazienti
adulti1, tendono a comparire nella terza decade di vita18.
Possono, tuttavia, essere presenti anche nell'infanzia12
e presentano una tendenza ad aumentare in numero e dimensione con
l'età1.
Risultati
cosmetici possono essere ottenuti mediante cauterizzazione o
laserterapia19.
Importante
è la diagnosi differenziale con altri tipi di lesioni
vascolari, in particolare gli angiomi stellati (spider naevi)
costituiti da una arteriola centrale da cui si irradiano numerosi
piccoli vasi; essi scompaiono alla digitopressione per poi
ricomparire con andamento centrifugo a dimostrazione dell'origine
arteriolare della lesione. Le localizzazioni tipiche sono a livello
del volto, parte alta del tronco ed estremità superiori,
raramente interessano le mucose; nell'infanzia è tipica la
localizzazione al dorso delle mani e delle dita. In età
adulta, diversamente da quella infantile, gli angiomi stellati sono
comunemente associati ad epatopatia cronica20.
Coinvolgimento gastrointestinale
Sanguinamenti
gastrointestinali (ematemesi o, più tipicamente, melena) si
verificano in meno di un terzo dei pazienti e generalmente
esordiscono dopo la quinta o sesta decade di vita21-24.
Sporadiche le segnalazioni in età pediatrica12.
Coinvolgimento
epatico
In più
del 30% dei soggetti adulti HHT è riportato un interessamento
asintomatico delle strutture vascolari del fegato.
Sono
limitate le segnalazioni di scompenso cardiaco, ipertensione portale
o patologia biliare secondarie a localizzazione epatica25.
Coinvolgimento
cerebrale
Malformazioni
artero venose a livello cerebrale sono riportate con una prevalenza
del 5-20% nella popolazione adulta e pediatrica. Il rischio di
emorragia cerebrale massiva è del 2-4% per anno nella
popolazione pediatrica affetta da HHT26.
Coinvolgimento
polmonare
Malformazioni
artero-venose (MAV) a livello polmonare sono complessivamente
riportate in una percentuale compresa tra il 5 e il 50% dei
pazienti27. La sintomatologia polmonare si manifesta
solitamente intorno alla seconda-terza decade di vita ed è
presente in meno di un terzo dei casi. I sintomi includono tosse,
dispnea e cianosi cutanea, specialmente periorale e ungueale, dovute
a ipossiemia arteriosa secondaria a shunt destro-sinistro. Il primo
segno clinico può tuttavia essere rappresentato da sequele
neurologiche di emorragie, embolie o ascessi cerebrali,
indipendentemente dalla presenza di sintomatologia polmonare.
Nonostante
le MAV possano essere presenti già alla nascita, rare sono le
segnalazioni di sintomatologia polmonare in età pediatrica
(15% dei casi)12, 28.
La
diagnosi si avvale della misurazione della saturazione dell'ossigeno
(in clino- e ortostatismo) associata ad indagini di imaging
(radiografia, tomografia computerizzata del torace con studio
angiografico e ecocardiografia).
In età
pediatrica il trattamento di prima scelta consiste
nell'embolizzazione percutanea della malformazione vascolare29.
Un follow-up clinico-strumentale dopo l'intervento è
necessario; utile inoltre la profilassi antibiotica in caso di
interventi chirurgici o cure dentarie.
Conclusioni
La malattia
di Rendu-Osler-Weber è una patologia difficile da
diagnosticare, specie in età infantile, sulla base dei soli
dati clinici30. Il sanguinamento nasale, pur essendo il
segno clinico più frequente e precoce, è comunque
comune in età pediatrica e pertanto, se isolato, aspecifico.
In un bambino con sintomatologia suggestiva di HHT è
fondamentale indagare la presenza di familiari con diagnosi certa di
HHT. In caso di dato anamnestico positivo è necessario
effettuare indagine molecolare; tale indagine dovrà essere
estesa a tutti i familiari al fine di identificare i soggetti affetti
e di avviarli ad un programma di follow-up, anche se ancora
asintomatici.
Il caso
descritto è emblematico di un esordio atipico e caratterizzato
da una sintomatologia totalmente differente da quella presentata dai
familiari affetti. Concludiamo pertanto sottolineando l'importanza
di un attenta anamnesi familiare che deve essere effettuata
ricordando l'estrema variabilità clinica intrafamiliare
caratteristica di questa patologia.
Bibliografia
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989;32:291-7.
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994;8:345-51.
- Ryder REJ, Mir MA, Freeman EA. An aid to the MRCP short cases. Oxford: Blackwell Scientific, 1986.
- Sutton HG. Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular system. Medical Mirror 1864;1:769-81.
- Legg W. A case of haemophilia complicated with multiple naevi. Lancet 1876:856-7.
- Rendu H. Epistaxis repetees chez un sujet porteur de petits angiomes cutanes et muquez. Gazette des Hopitaux Civils et Militaires (Paris) 1896;135:1322-3.
- Osler W. On a family form of recurring epistaxis, associated with multiple telangiectases of the skin and mucous membranes. Bulletin of the Johns Hopkins Hospital 1901;12:333-7.
- Weber F. Multiple hereditary developmental angiomata (telangiectases) of the skin and mucous membranes associated with recurring haemorrhages. Lancet 1907:160-2.
- Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997;61:60-7.
- Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91:66-7.
- Porteus MEM, Burn J, Proctor SJ. Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet 1992;29:527-30.
- Folz BJ, Zoll B, Alfke H, Toussaint A, Maier RF, Werner JA. Manifestations of hereditary hemorrhagic telangiectasia in children and adolescents. Eur Arch Otorhinolaryngol 2006;263:53-61.
- Guttmacher AE, Marchuk DA, White RI Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995;333:918-24.
- Saunders WH. Septal dermoplasty for hereditary telangiectasia and other conditions.Otolaryngol Clin North Am 1973;6:745-55.
- Parkin JL, Dixon JA. Laser photocoagulation in hereditary hemorrhagic telangiectasia. Otolaryngol Head Neck Surg 1981;89:204-8.
- Harrison DF. Use of estrogen in treatment of familial hemorrhagic telangiectasia. Laryngoscope 1982;92:314-20.
- Elden L, Montanera W, Terbrugge K, Willinsky R, Lasjaunias P, Charles D. Angiographic embolization for the treatment of epistaxis: a review of 108 cases. Otolaryngol Head Neck Surg 1994;111:44-50.
- Braverman IM, Keh A, Jacobson BS. Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J Ivest Dermatol 1990;95:422-7.
- Morelli JG, Huff JC, Weston WL. Treatment of congenital telangiectatic vascular malformations with thepulsed-dye laser (585 nm). Pediatrics 1993;92:603-6.
- Finn SM, Rowland M, Lawlor F, Kinsella W, Chan L, Byrne O, O'Mahony O, Bourke B. The significance of cutaneous spider naevi in children. Arch Dis Child. 2006;91:604-5.
- Vase P, Grove O. Gastrointestinal lesions in hereditary hemorrhagic telangiectasia. Gastroenterology. 1986;91:1079-83.
- Korzenik JR. Hereditary hemorrhagic telangiectasia and other intestinal vascular anomalies. Gastroenterologist 1996;4:203-10.
- Sharma VK, Howden CW. Gastrointestinal and hepatic manifestations of hereditary hemorrhagic telangiectasia. Dig Dis 1998;16:169-74.
- Abdalla SA, Geisthoff UW, Bonneau D, Plauchu H, McDonald J, Kennedy S, et al. Visceral manifestations in hereditary haemorrhagic telangiectasia type 2. J Med Genet 2003;40:494-502.
- Garcia-Tsao G, Korzenik JR, Young L, Henderson KJ, Jain D, Byrd B, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J 2000;343:931-6.
- Morgan T, McDonald J, Anderson C, Ismail M, Miller F, Mao R, et al. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome). Pediatrics 2002;109:E12.
- Kjeldsen AD, Oxhoj H, Andersen PE, Green A, Vase P. Prevalence of pulmonary arteriovenous malformations (PAVMs) and occurrence of neurological symptoms in patients with hereditary haemorrhagic telangiectasia (T). J Intern Med 2000;248:255-62.
- Olgunturk R, Oguz D, Tunaoglu S, Ikizler C, Sezgin A, Kula S. Pulmonary arteriovenous fistula in the newborn: a case report of Rendu-Osler-Weber syndrome and a review of the literature. Turk J Pediatr 2001;43:332-7.
- Faughnan ME, Thabet A, Mei-Zahav M, Colombo M, Maclusky I, Hyland RH, et al. Pulmonary arteriovenous malformations in children: outcomes of transcatheter embolotherapy. J Pediatr 2004;145:826-31.
- Giordano P, Nigro A, Del Vecchio GC, Sabbà C, De Mattia D. HHT in childhood: screening for special patients. Curr Pharm Des. 2006;12:1221-5.
Vuoi citare questo contributo?