Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Maggio 2006 - Volume IX - numero 5
M&B Pagine Elettroniche
Pediatria per l'ospedale
I
meccanismi della proteinuria (parte prima)
Membro
della Commissione Nazionale Vaccini
Indirizzo
per corrispondenza: bartolozzi@unifi.it
Le
forme ereditarie di proteinuria comprendono un gruppo eterogeneo di
malattie renali, nelle quali predominano i segni e i sintomi della
disfunzione glomerulare e della proteinuria. Gli approfondimenti in
questo settore hanno permesso di conoscere meglio i meccanismi della
filtrazione glomerulare e le cause della proteinuria. Una volta di
più gli esperimenti spontanei della natura ci permettono di
conoscere i più intimi meccanismi della fisiologia.
Un
articolo di revisione di tutti questi aspetti della fisiopatologia
renale è comparso di recente in letteratura: Tryggvason K,
Patrakka J, Wartiovaara. Hereditary proteinuria syndromes and
mechanisms of proteinuria. N Engl J Med 2006;354:1387-401). La
pubblicazione comprende 106 voci bibliografiche; gli utori lavorano
al Karolinska Institute di Stoccolma e all'Istituto di
Biotecnologia dell'Università di Helsinki.
Come
sarà possibile rilevare dalla lettura, la funzionalità
della filtrazione glomerulare è divenuta oggi quanto mai
complessa; la difficoltà risiede soprattutto nell'introduzione
di una nuova terminologia. La revisione verrà suddivisa in due
parti.
Ildecorso di questo gruppo di malattie può essere il più
diverso:
- Alcuni pazienti si presentano con grave proteinuria e con il quadro della sindrome nefrosica congenita.
- Altri presentano solo una moderata proteinuria e una glomerulosclerosi focale segmentale.
Senza
tener conto delle diverse cause che sono in gioco, si può dire
che la malattia spesso evolve in modo progressivo per arrivare
allo stadio terminale della malattia renale. La classificazione di
queste sindromi è sempre stata difficile, perché
variano sia per l'età d'inizio che per le manifestazioni
cliniche, ma negli ultimi anni sono stati fatti notevoli progressi
nel determinare le cause genetiche di queste sindromi. Per
prima cosa è stato visto che le mutazioni dello stesso gene
possono portare sia a una sindrome nefrosica congenita che a
una glomerulosclerosi focale segmentale. Oggi tutte queste
malattie vengono classificate come sindromi della proteinuria
ereditaria. Da un punto di vista clinico è importante sapere
che alcune sindromi da proteinuria ereditaria rispondono alla
terapia, mentre altre no. Per questa ragione, le determinazioni
genetiche, che sono disponibili per alcune delle sindromi ereditarie,
vanno eseguite, quando questo sia possibile. Le conoscenze dei
meccanismi della filtrazione glomerulare e della proteinuria sono
ancora poco diffuse, ma questo campo particolare è attualmente
sottoposto a ricerche intense e produttive.
La
barriera della filtrazione glomerulare
Le cause
principali delle sindromi della proteinuria ereditaria risiedono in
alterazioni della barriera della filtrazione glomerulare della
corteccia renale (vedi Figura IA e IB).
Questa barriera ha tre strati:
- L'endotelio fenestrato;
- La membrana basale glomerulare;
- I podociti, insieme a un diaframma fenestrato fra i processi dei piedi dei podociti, detti comunemente pedicelli. (vedi Figura IC e ID)
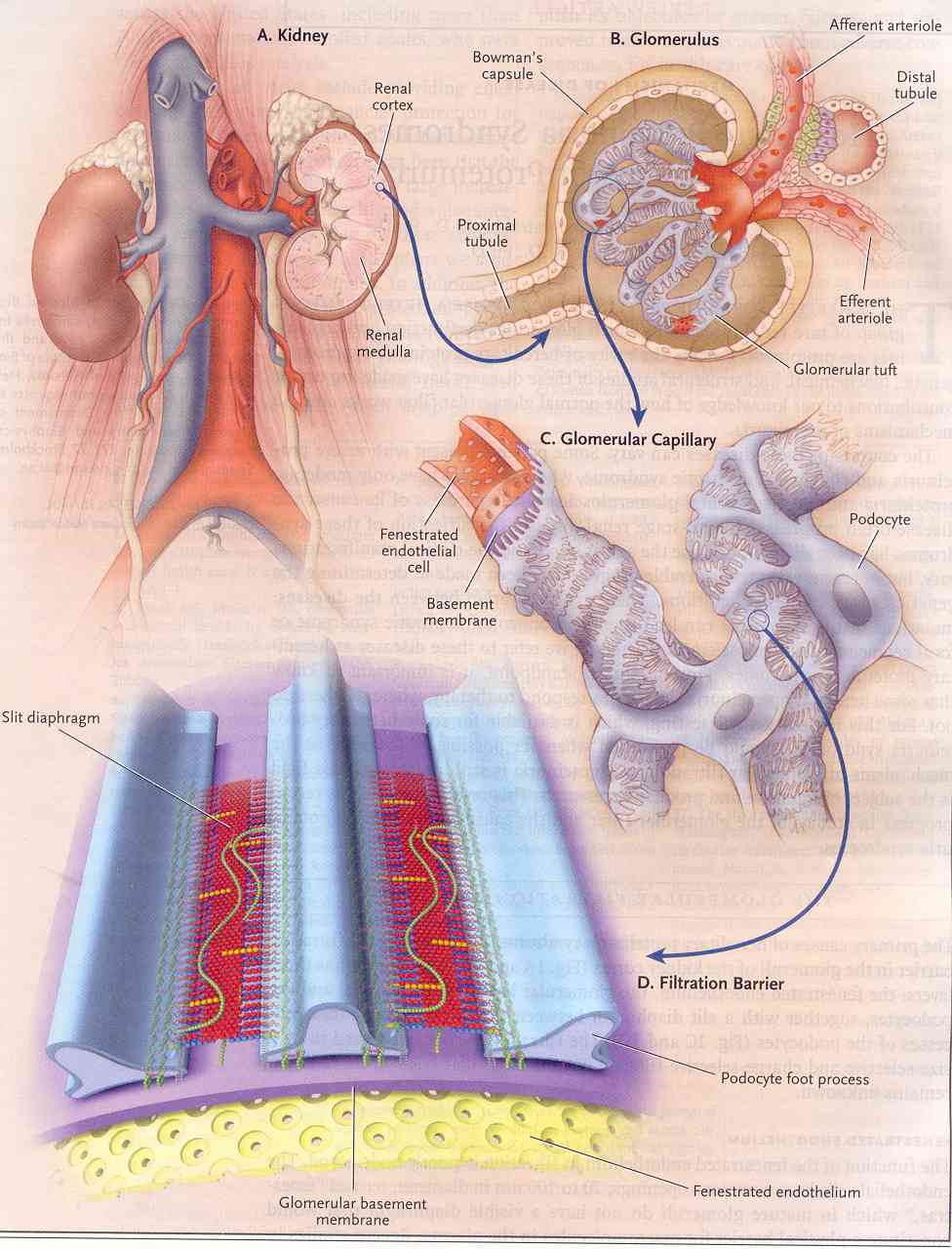
Figura
1. Filtrazione glomerulare
Ogni
rene contiene circa 1 milione di glomeruli nella corteccia renale
(Figura 1A). Nel riquadro B si vede un'arteriola afferente, che
entra nella capsula di Bowman e si sfiocca in molti capillari, che
formano il gomitolo glomerulare. Le pareti dei capillari
costituiscono il filtro. Il filtrato del plasma (urina primitiva)
passa nel tubulo prossimale, mentre il sangue non filtrato ritorna
nella circolazione generale, attraverso l'arteriola efferente (il
glomerulo è una rete mirabile arteriosa). La barriera di
filtrazione della parete del capillare è costituita da un
endotelio fenestrato, dalla membrana basale glomerulare e dallo
strato dei processi pedicelli dei podociti interdigitanti (riquadro
C). Nel riquadro D, una sezione trasversale del capillare glomerulare
rappresenta lo strato endoteliale fenestrato e la membrana basale
glomerulare con i processi pedicelli dei sovrastanti podociti. Un
diaframma ultrasottile fenestrato presenta finestre di filtrazione
fra i pedicelli dei podociti, leggermente al di sopra della membrana
basale.
Per
mostrare il diaframma fenestrato, in questa figura i processi
pedicellari dei podociti sono più piccoli.
La
barriera di filtrazione si pensa sia legata alla grandezza e alla
carica elettrica del filtro, ma la base molecolare della sua funzione
rimane ancora sconosciuta.
Endotelio
fenestrato
La
funzione dell'endotelio fenestrato nella filtrazione è poco
conosciuta. Le cellule endoteliali a questo livello hanno numerose
aperture di 70-100 nm di diametro, chiamate finestre, che nel
glomerulo maturo non rappresentano un diaframma che costituisca una
barriera fisica per le macromolecole presenti nel plasma.
Il
fattore di crescita dell'endotelio vascolare, derivato dai
podociti, ha un ruolo importante nello sviluppo dell'endotelio e
nel mantenimento della sua fenestrazione. Le cellule dell'endotelio
glomerulare hanno un glicocalice sulla loro superficie, contenente le
sialoproteine e i proteoglicani a cariche negative, ma non ci sono
prove che il glicocalice giochi un ruolo nella filtrazione.
Membrana
basale glomerulare
La
membrana basale glomerulare è una matrice acellulare con uno
spessore di 300-350 nm, che fornisce un sopporto strutturale alla
parete dei capillari. Il suo principale componente è il
collageno tipo IV, insieme ai proteoglicani, alla laminina e al
nidogeno. Nel feto, le molecole del collageno tipo IV a triploelica
della membrana basale glomerulare contengono catene a1 (IV) e a2 (IV)
in un rapporto 2:1; questa forma di collageno è più
tardi rimpiazzata da molecole tipo adulto
contenenti
a3 (IV), a4 (IV) e a5 (IV), con un rapporto 1:1:1. La fitta rete del
collageno tipo IV fornisce forza elastica alla membrana, ma
probabilmente non contribuisce a selezionare le molecole per
grandezza o per carica elettrica a livello del filtro glomerulare.
Questo punto di vista è confermato dal reperto che mutazioni
del collageno IV tipo adulto portano a distorsioni della struttura
della membrana basale glomerulare in pazienti con sindrome di Alport,
che presentano ematuria come manifestazione renale, ma in genere
presentano solo lieve proteinuria.
Studi con
il microscopio elettronico hanno identificato siti anionici nella
membrana basale glomerulare. Questi siti si pensa siano localizzati
sulle catene laterali di eparan solfato e condroitin solfato del
perlacan e dell'agrin. Le cariche anioniche sembra siano importanti
per la filtrazione, poiché la rimozione o la riduzione
enzimatica nel numero della cariche si accompagna a proteinuria.
Le
laminine sono grandi proteine eterotrimeriche, importanti per la
differenziazione e per l'adesione cellulare. Esse hanno una
funzione strutturale: assemblano se stesse in una rete di laminina in
molti tipi di membrana basale. Nella membrana basale glomerulare del
feto, una forma iso della laminina (la laminina 10) è
rimpiazzata dopo la nascita dalla laminina 11. La mancanza
sperimentale del gene β2 della laminino 11, causa proteinuria e
morte neonatale. Di recente è stato dimostrato che mutazioni
del gene della laminino β2, determina la sindrome di Pierson, una
forma precoce e letale della sindrome nefrosica congenita. La
laminina 11 è quindi indispensabile per la funzione della
membrana basale glomerulare.
Il
diaframma fenestrato e i podociti
Il
diaframma fenestrato ha un ruolo importante e diretto nella
filtrazione glomerulare. Alcune delle sue componenti proteiche sono
interessate nel meccanismo della proteinuria. Queste proteine formano
un complesso che contribuisce alla struttura del diaframma
fenestrato, connette il diaframma con il citoscheletro intracellulare
di actina e partecipa alla segnalazione, relativa al turnover del
filtro glomerulare. La maggiore parte di queste proteine sono
essenziali per la funzione del diaframma fenestrato e per la
filtrazione glomerulare, poiché mutazioni o attivazioni dei
rispettivi geni causa proteinuria.
Queste
proteine sono rappresentate dalla nefrina, dal Neph 1 e Neph 2, dal
FAT1 e FAT2, dalla podocina, dal CD2AP e da altre.
Nefrina
La
nefrina è stata la prima proteina del diaframma fenestrato a
essere identificata. Il gene della nefrina è mutato nella
sindrome nefrosica congenita di tipo finlandese (CNF o sindrome
nefrosica tipo 1). Nel rene soltanto i podociti formano la nefrina:
l'inattivazione del gene della nefrina causa una proteinuria
massiva nell'animale da laboratorio, assenza di un diaframma
fenestrato e la morte neonatale. La nefrina ha un corto dominio
intracellulare, un dominio transmembrana e un dominio extracellulare
con 8 elementi distali IgG-simili e un elemento prossimale
fibronectina III-simile (vedi Figura 2A). Le
molecole di nefrina interagiscono l'una con un'altra in modo
omofilico. La lunghezza del dominio extracelllulare è di circa
35 nm e le molecole di nefrina, originate dagli adiacenti processi
podocitici, si pensa interagiscano con la parte di mezzo della
finestra per formare una struttura filtrante (vedi Figura
2B). L'importanza della fosforilazione Fyn-dipendente della
nefrina (Fyn è un membro della famiglia Src della proteina
tirosina chinasi) è sottolineata dal fatto che negli animali
da esperimento mancanti di Fyn chinasi si manifestano proteinuria e
assenza dei podociti.
Neph1
e Neph2
Neph1 e
Neph2 sono proteine, strutturalmente in relazione con la nefrina:
ognuna è formata da 5 motivi extracellulari IgG-simili (vediFigura 2C). Esse appartengono a una famiglia
di proteine transmembrana (Neph1, Nepoh2 e Neph3, chiamate anche
filtrino) che si trovano in molti tessuti. Neph1 e Neph2 sono
localizzate nel diaframma fenestrato. Quando siano fosforilate queste
proteine partecipano alle segnalazioni intracellulari. Gli animali
deficienti di Neph 1 hanno proteinuria e muoiono nelle prime 8
settimane di vita; il significato funzionale di Neph 2 e di Neph 3 è
sconosciuto.
FAT1
e FAT2
FAT1 e
FAT2 sono proteine transmembrana molto grandi del diaframma
fenestrato, costituite da 34 segmenti uguali caderina-simili (vediFigura 2E) (La caderina appartiene alla
famiglia di molecole di adesione cellulare, calcio-dipendenti). La
mancanza di FAT1 nel topo causa la perdita del diaframma fenestrato e
proteinuria, la mancanza del cervello primitivo e difetti oculari;
segue la morte in epoca perinatale. La mancanza di FAT2 causa solo
proteinuria.
P-caderina
e la molecola 4 di adesione giunzionale sono state identificate nel
diaframma fenestrato, ma queste ultime proteine non sono
indispensabili alla filtrazione glomerulare, per cui il loro ruolo
deve ancora essere chiarito.
Podocina
Alla
ricerca del gene mutato nella sindrome nefrosica congenita
cortico-resistente è stata trovata la podocina, una proteina
localizzata solo nella regione del diaframma fenestrato. Essa è
una proteina di membrana a forma di forcina per capelli, con ambedue
le estremità dirette verso lo spazio intracellulare (vediFigura 2F). La podocina interagisce con i
domini intracellulari della nefrina e del Neph1 e con la proteina
associata al CD2 (CD2AP). Nei topi privi di podocina si sviluppa una
grave proteinuria, per cui essi muoiono dopo pochi giorni dalla
nascita.
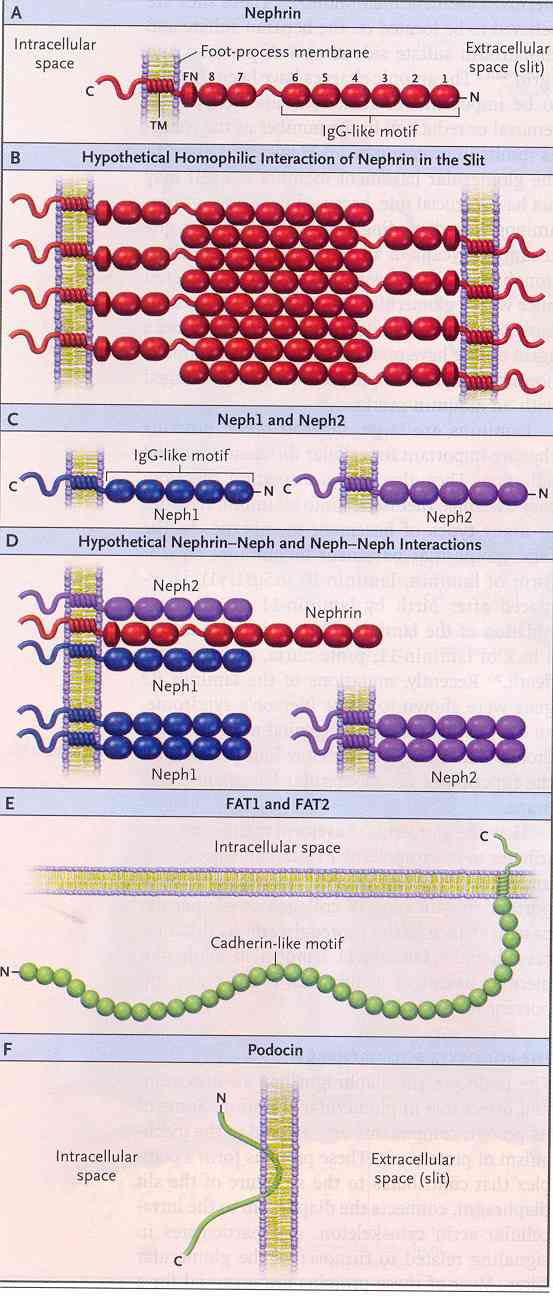
Figura
2. Componenti del complesso proteico del diaframma fenestrato nei
processi dei pedicelli dei podociti.
Come
si osserva nel segmento A, la nefrina ha un corto dominio
intracellulare, un dominio transmembrana (TN) e un dominio
N-terminale extracellulare con un elemento prossimale di fibronectina
tipo III-simile e 8 elementi IgG-simili, che vengono numerati a
partire dall'N terminale. Nel segmento B si osservano le
interazioni omofiliche fra le molecole di nefrina. Nello spazio
extracellulare, le molecole dei processi podocitici adiacenti si
pensa interagiscano con il centro della finestra per formare la spina
dorsale del diaframma fenestrato. Questo tipo di assemblamento può
permettere ai pori di essere localizzati su ambedue i lati della
parte centrale densa. Nel segmento C si osserva che ognuna delle
molecole transmembrana Neph1 e Neph2 contiene 5 motivi extracellulari
IgG simili. Come si vede nel segmento D, le molecole Neph si pensa
abbiano interazioni omofiliche con le specole adiacenti di nefrina.
Come appare nel segmento E, FAT1 e FAT 2 sono proteine transmembrana
di più di 500 kD, che contengono 34 consecutivi elementi
extracellulari caderina-simili. Il loro modo di interagire con le
altre proteine della membrana fenestrata non è stato ancora
caratterizzato. Nella parte F si osserva che la podocina è una
proteina integrale di membrana di circa 30 kD, con i suoi terminali N
e C localizzati all'interno della cellula.
CD2AP
CD2AP è
una proteina intracellulare, inizialmente caratterizzata come una
proteina "adattatore" del linfocita T CD2. La maggior parte
dei topi, privi di questa proteina, muoiono per una sindrome
nefrosi-simile a 6-7 settimane di vita; la proteina è
localizzata nelle regione dei podociti del diaframma fenestrato del
glomerulo. Persone che siano eterozigoti per un allele CD2AP
difettoso hanno un fenotipo renale complesso: polimorfismi del gene
umano si associano allo sviluppo di una glomerulonefrite e di una
glomerulosclerosi. Perciò il CD2AP può essere
considerato come un gene della suscettibilità alla
glomeruolonefrite. Il CD2AP può interagire con i domini
intracellulari della nefrina e della podocina; la proteina è
stata associata anche con l'endocitosi. Il CD2AP è
interessato inoltre alle segnalazioni nel diaframma fenestrato.
Altre
proteine costituenti il diaframma fenestrato
ZO-1 è
una proteina intracellulare, ampiamente espressa, connessa con le
giunzioni rigide epiteliali; essa è localizzata nella regione
del diaframma fenestrato e può interagire con le proteine
della famiglia Neph. Il ruolo della proteina ZO-1 nel complesso delle
proteine del diaframma fenestrato non è conosciuto.
La
scoperta di specifici componenti del complesso proteico del diaframma
fenestrato ha portato a nuove conoscenze nella fisiologia della
barriera di filtrazione e dei meccanismi della proteinuria. Il fatto
che la maggioranza di queste proteine sia fondamentale per lo
sviluppo e la funzione normale del rene, sottolinea l'importanza
del diaframma fenestrato nel determinare le caratteristiche di
filtrazione del glomerulo.
Struttura
del diaframma fenestrato
Il
diaframma fenestrato (vedi Figura 3A e 3B) ha una vera struttura di
filtro poroso ?
Sulla
base dei vecchi reperti alla microscopia elettronica, è stato
proposto che il diaframma fenestrato sia una struttura ordinata,
simile a una chiusura lampo, con pori, più piccoli come
diametro delle molecole di albumina. Ma recenti analisi del diaframma
fenestrato con una nuova metodica di microscopia elettronica hanno
dimostrato che questo strato sottile contiene parti convolute che
attraversano la linea mediana della finestra di filtrazione e spesso
formano lamine con pori del diametro della molecola di albumina o più
piccoli, localizzati su ambedue i lati della densità centrale
(vedi Figura 3C). La microscopia
immuno-elettronica e la tomografia elettronica sono state usate per
evidenziare che gli elementi IgG1 e IgG2 della nefrina sono nella
regione centrale del diaframma fenestrato (vedi Figura
4A, 4B e 4C). D'altra parte le molecole di nefrina
immuno-marcate in soluzione (vedi Figura 4D)
assomigliano a una classe di componenti del diaframma fenestrato,
dimostrati con lo stesso metodo.
Tutto
questo suggerisce che le proteine del diaframma fenestrato formano
una struttura chiusura lampo-simile con un'ampiezza costante di 40
nm (vedi Figura 5). L'esatta localizzazione
e l'interazione della Neph1, Neph, FAT1 e FAT2 con queste
proteine non è ancora conosciuta. Queste proteine
interagiscono all'interno della cellula con molte altre proteine
che si connettono con il citoscheletro o partecipano ai segnali
cellulari.
Se, come
sembra, il diaframma fenestrato è un vero filtro, selettivo
per grandezza, c'è da chiedersi perché esso non si
ostruisca. Non c'è una chiara risposta a questa domanda, ma
è possibile che le cariche negative dei glicoaminoglicani,
presenti nella membrana basale glomerulare e sulla superficie dei
podociti, mediante un effetto di gel-esclusione o con qualche altro
meccanismo non identificato fino a ora, respingano le proteine dal
diaframma fenestrato in modo tale da prevenire l'ostruzione.
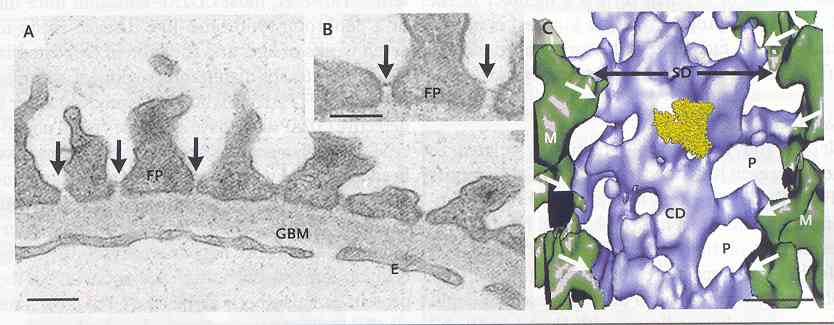
Figura
3. Immagini alla microscopia elettronica (parte A e B) e immagini
elettrono-tomografiche (parte C) del diaframma podocitico fenestrato.
Nella
parte A, una sezione del capillare glomerulare umano mostra le
finestre di filtrazione del diaframma fenestrato (frecce) fra i
processi pedicellari dei podociti (FP). Si vedono la membrana basale
glomerulare (GBM) e una cellula endoteliale (E). La linea della scala
è lunga 250 nm.
La
parte B mostra il diaframma fenestrato (freccia) a un ingrandimento
maggiore. La linea della scala è lunga 150 nm.
La
parte C mostra una tomografia elettronica sottile a tre dimensioni
del diaframma fenestrato di un topo (SD), visto dal davanti. Parti
trasversali (frecce) si estendono dalla membrana di superficie dei
podociti (M) alle parti centrali dense (CD), formando dei pori (Ps).
Il tomogramma offre una ricostruzione di una superficie. Per
confrontare il diametro dei pori, è stata superimposta in
giallo una molecola cristallina di sieroalbumina. La linea della
scala è lunga 10 nm.
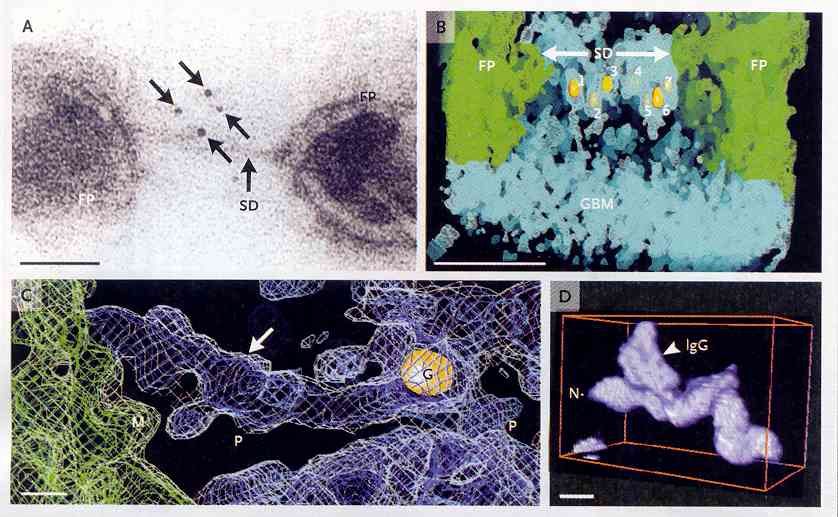
Figura
4. Immunomarcatura della nefrina nel diaframma fenestrato umano e
nefrina ricombinante in soluzione.
Nella
parte A è mostrata una micrografia elettronica della nefrita,
marcata con oro (le frecce indicano la parte N terminale IgG 1 e 2
della nefrina) in un diaframma fenestrato, tagliato obliquamente
(SD), fra due processi podocitici (FP). La linea della scala è
lunga 40 nm.
Nella
parte B, un tomogramma mostra una finestra di filtrazione con
all'esterno la membrana basale glomerulare (GMB), i processi
pedicillari e il diaframma fenestrato. I numeri da 1 a 7 indicano le
parti, marcate con oro, della nefrina sotto il diaframma fenestrato.
La linea della scala è lunga 40 nm.
Nella
parte C si vede il primo piano di una parte trasversale (freccia) di
circa 35 nm di lunghezza nel diaframma fenestrato. Sono visibili
anche i pori (P). Si vede il terminale N della nefrina, marcato con
oro, all'estremo distale della parte trasversale. La linea della
scala è lunga 5 nm.
La
parte D mostra un tipico elemento di nefrina umana ricombinante
extracellulare, di circa 35 nm di lunghezza, in una soluzione
contenente IgG anti-nefrina umana. La lunghezza della scala è
di 5 nm.
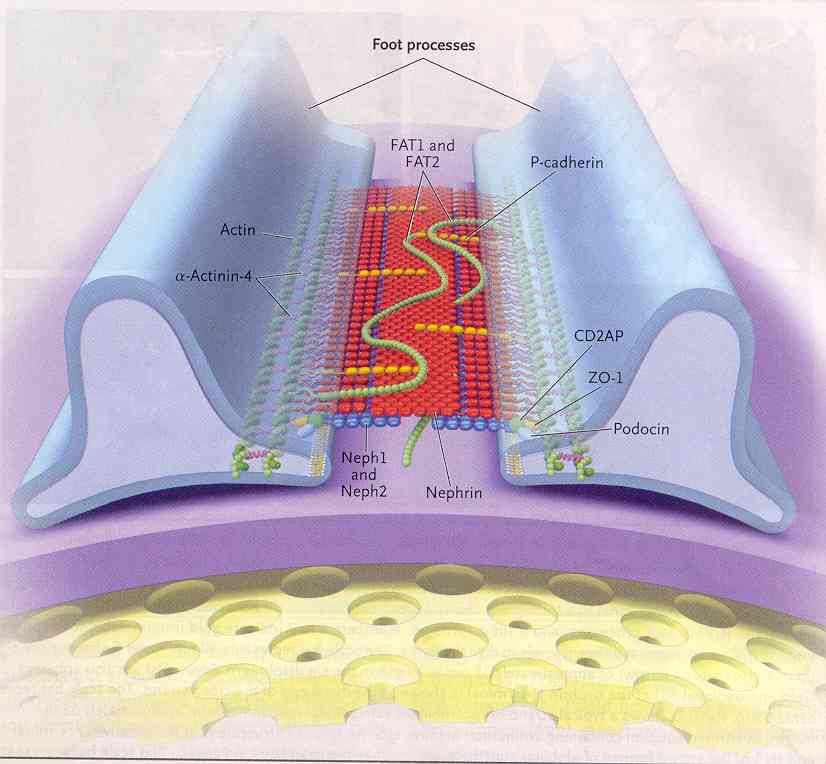
Figura
5. Componenti del complesso proteico del diaframma fenestrato che
forma un filtro poroso.
Le
molecole di nefrina dall'altro lato dei piedi dei dopociti
interagiscono al centro della finestra, formando una densità
centrale con pori da ambo i lati. La struttura cerniera-simile
formata dalle molecole di nefrina probabilmente mantiene lo spessore
della finestra a circa 40 nm. La nefrina interagisce anche con altre
proteine nella finestra, come FAT1 e FAT2. Le molecole più
corte Neph1 e Neph2 possono interagire con altre proteine, come anche
con la parte prossimale delle molecole di nefrina, per stabilizzare
la struttura del diaframma fenestrato. La nefrina e le molecola Neph
interagiscono con la podocina intracellulare e con la proteina
associata CD2. Questa presumibilmente connette il complesso delle
proteine del diaframma fenestrato con ZO-1 e con gli elementi di
actina. Gli elementi di actina sono uniti alle molecole di
a-actina-4.
Vuoi citare questo contributo?