Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Gennaio 2006 - Volume IX - numero 1
M&B Pagine Elettroniche
Pediatria per l'ospedale
ß-talassemia
(Parte
prima)
Membro
della commissione nazionale vaccini
Indirizzo
per corrispondenza:bartolozzi@unifi.it
La
ß-talassemia è una malattia, su base ereditaria, causata
da un difetto quantitativo nella produzione di emoglobina. In pratica
essa è causata da una diminuzione nella produzione di catene ß
dell'emoglobina, che colpisce gran parte degli organi e si associa
a un'elevata morbilità e letalità.
E'
diffusa in tutto il mondo e il nostro Paese, insieme alla Grecia, è
quello più gravemente colpito: si pensa che in Italia vi siano
circa 2 milioni di portatori della tara talassemica.
La
malattia, allo stato omozigote, venne riconosciuta a Chicago da
Cooley (di cui porta il nome) fra gli italiani emigrati negli Stati
Uniti. Una recente revisione (Rund D, Rachmilewitz E.
ß-talassemia. N Engl J Med 2005, 353:1135-46), completa e
aggiornatissima, offre l'occasione per tornare a parlare di questa
importante malattia.
La
talassemia è l'alterazione genetica più diffusa nel
mondo: il 4,84% della popolazione mondiale presenta una variante
dell'emoglobina, mentre 1.67% è eterozigote per la
ß-talassemia e la ß-talassemia, l'1,92% è
portatore dell'emoglobina S (drepanocitosi), lo 0,95%
dell'emoglobina E e lo 0,29% dell'emoglobina C. Da questo
consegue che un bambino che nasca con alterazioni dell'emoglobina,
compresi gli omozigoti e gli eterozigoti per ß-talassemia e la
ß-talassemia, corrisponde al 2,4 per 1000 nascite, di cui
l'1,96 ha falcizzazione (emoglobina S) e lo 0,44 ha una talassemia.
Alterazioni
molecolari e cellulari
La
ß-talassemia è causata da una delle oltre 200 mutazioni
puntiformi e raramente da una delezione. Poiché di essa fanno
parte molte alterazioni genetiche, che colpiscono in modo diverso la
sintesi delle catene globiniche , il quadro clinico della talassemia
è clinicamente molto eterogeneo. Tuttavia i soli fattori
genetici non sono sufficienti per spiegare l'evidente variabilità
clinica, per cui si ritiene che esistano altri fattori che agiscano a
carico di altri geni: la differenza fra genotipi e fenotipi è
particolarmente evidente nella talassemia intermedia e nella
talassemia con emoglobina E (si tratta di un'emoglobina, frequente
soprattutto in Tailandia, in cui nella ?-catena una lisana è
stata sostituita dall'acido glutammico in posizione 26).
L'emolisi
e l'eritropoiesi inefficace causano ambedue l'anemia, presente
nella talassemia: questi due meccanismi patogenetici differiscono
come intensità nelle diverse forme di anemia.
Il
midollo osseo dei pazienti con talassemia contiene da 5 a 6 volte il
numero di eritroblasti che si trova nel midollo di soggetti sani,
mentre il numero delle cellule in apoptosi è 15 volte più
alto del normale negli eritroblasti appartenenti allo stadio
policromatofilo e ortocromatico. L'accelerata apoptosi, la causa
principale dell'eritropoiesi inefficace, è determinata dalla
presenza in eccesso di depositi di ?-catene negli eritroblasti.
L'accelerazione dell'apoptosi è associata a un aumento
dell'esposizione extracellulare alla fosfatidilserina, un
importante segnale per la rimozione della cellula da parte dei
macrofagi attivati, il cui numero è aumentato nel midollo
osseo dei talassemici (vedi figura
1).
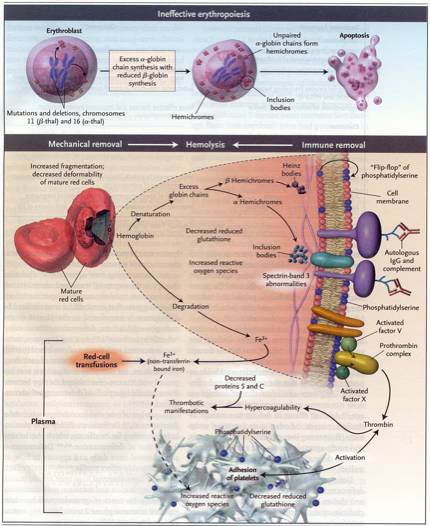
(Rund
D, Rachmilewitz E, 2005).
L'ossidazione
dell'emoglobina alfa porta alla formazione di emicromi, il cui
tasso di formazione determina il tasso di emolisi. Poiché gli
emicromi di alfa catene si formano più facilmente di quelli di
catene beta o gamma, la betatalassemia è clinicamente molto
più grave della alfa-talassemia. Gli emicromi si legano e
modificano molti componenti della membrana del globulo rosso maturo.
Dopo la precipitazione degli emicromi e la disintegrazione dell'eme,
sono liberati composti contenenti ferro tossico, non legato alla
transferrina. Il ferro libero catalizza la formazione di comoposti di
ossigeno attivo. L'ossidazione da parte del ferro delle proteine
della membrana e la formazione di antigeni della senescenza del
globulo rosso, come la fosfatidilserina, rendono rigido e deformato
il globulo rosso talassemico, che si aggrega e viene rimosso
prematuramente. La fosfatidilserina è anche coinvolta
nell'attivazione del sistema della coagulazione.
Manifestazioni
cliniche e trattamento
Anemia
e terapia trasfusionale
Per
mantenere e migliorare la crescita e lo sviluppo del bambino è
necessario eseguire una terapia trasfusionale regolare per tenere i
livelli di emoglobina almeno al di sopra dei 9-10 g/dL: in tal modo
si riduce l'epatosplenomegalia, dovuta anche all'eritropoiesi
extramidollare, e le deformità ossee.
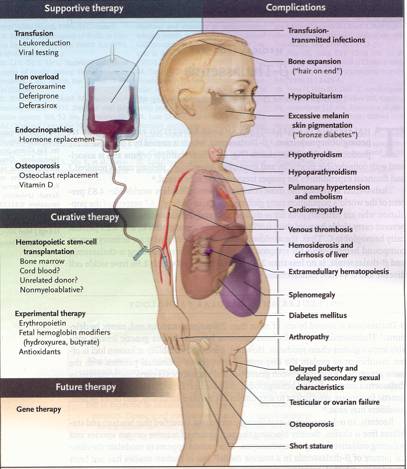
Endocrinopatie
e malattie ossee
Alterazioni
della crescita ed endocrinopatie, specialmente ipogonadismo, sono
aspetti comuni della talassemia (figura
2).
Le
manifestazioni dell'anemia cronica e quelle del carico di ferro
sono più comuni nei pazienti di età più avanzata
o in quelli nei quali la terapia chelante è stata
insufficiente. Il trattamento con ormone della crescita ha avuto un
risultato variabile. L'ipogonadismo ipogonadotropo determina
l'infertilità, ma può essere corretto con l'uso di
preparati ormonali nei soggetti di sesso maschile. Un piccolo numero
di soggetti di sesso femminile, inclusi quelli con talassemia major o
talassemia intermedia, sono capaci di entrare in stato di gravidanza,
sia spontaneamente (grazie a un'adeguata terapia chelante) sia con
l'applicazione di tecniche di riproduzione assistita. La gravidanza
in genere si svolge normalmente, se la funzione cardiaca è
buona.
Considerevoli
conseguenze nei pazienti più avanti con l'età sono
legate all'osteopenmia e all'osteoporosi, che spesso si
accompagnano a dolori ossei e a fratture. La loro patogenesi è
complessa e multifattoriale. L'espansione del midollo osseo, dovuta
all'eritropoiesi inefficace, alle alterazioni endocrine e alla
complicazioni, legate al trattamento, è un fattore importante
nella genesi della sofferenza dell'osso. Una vigorosa chelazione si
associa spesso a displasia ossea, associata a deferoxamina, tanto da
rallentare la velocità di crescita nei bambini: essa può
essere solo parzialmente reversibile.
Il
trattamento delle lesioni ossee richiede un accurato monitoraggio
della chelazione, una regolamentazione degli stili di vita (aumentato
introito di calcio, attività fisica e completa astensione dal
fumo), la terapia ormonale e la somministrazione di vitamina D. La
somministrazione di bifosfonati, inibitori degli osteoclasti,
determina una riduzione del riassorbimento dell'osso e può
meritare di essere provata, ma sono necessari ulteriori studi prima
che essa entri nell'uso routinario.
Sovraccarico
di ferro, patogenesi, valutazione e terapia
Il
sovraccarico di ferro è il principale responsabile della
letalità e della morbilità per talassemia. I depositi
di ferro avvengono nei visceri (principalmente nel cuore, nel fegato
e nelle ghiandole endocrine), determinando un danno tissutale e
infine una disfunzione e un'insufficienza d'organo (figura
2). Le conseguenze cardiache infine sono la causa principale di
morte. Vi contribuiscono il sovraccarico trasfusionale di ferro e
l'eccessivo assorbimento intestinale. E' infatti un paradosso che
continui un eccessivo assorbimento di ferro dall'intestino,
nonostante il forte aumento del ferro totale nell'organismo.
L'epcidina
è un piccolo peptide che inibisce l'assorbimento di ferro
nel tenue. I livelli di epcidina aumentano di regola quando i
depositi di ferro sono elevati. Ma i livelli di epcidina sono
inappropriatamente bassi nei pazienti con talassemia intermedia e in
quelli con talassemia major. E' probabile che la presenza di un
fattore umorale inibisca l'RNA messaggero dell'epcidina. Questi
rilievi suggeriscono che la somministrazione di epcidina o di agenti
che ne aumentino l'attività sarebbe utile per inibire
l'assorbimento del ferro.
(Rund
D, Rachmilewitz E, 2005).
L'anemia,
dovuta alla talassemia, può essere grave: essa si accompagna a
eritropoiesi inefficace, con espansione ossea ed ematopoiesi
extramidollare nel fegato, nella milza e in altre sedi (in sede
paravertebrale). La terapia con le trasfusioni, che rappresenta il
trattamento principale, porta a uno sviluppo normale e sopprime
l'eritropoiesi inefficace. Il carico di ferro deriva sia dalla
emosiderosi trasfusionale, che dall'eccessivo assorbimento di ferro
dall'intestino. Il ferro si deposita nel cuore, nel fegato, e nelle
ghiandole endocrine, determinando danni evidenti a carico di questi
organi, con variabile insufficienza ghiandolare. La più grave
conseguenza del carico di ferro si manifesta a carico del cuore, con
una forte cardio-tossicità, per la quale si rende necessaria
la chelazione. La talassemia può essere curata con il
trapianto di midollo osseo. Nel futuro saranno disponibili altre
terapia (modificatori dell'emoglobina fetale, antiossidanti,
terapia genica e altri trattamenti molecolari).
Misurazioni
accurate dei depositi di ferro sono essenziali per la valutazione e
la conduzione della terapia chelante. L'indicatore migliore è
la determinazione della ferritina: livelli di ferritina al di sotto
dei 2.500 ng/mL si associano a un miglioramento della sopravvivenza,
senza complicazioni cardiache. Ma è difficile raggiungere
questi livelli, quando vi sia una malattia del fegato. Tralasciando
la biopsia del fegato perché invasiva, oggi si preferisce la
misurazione dei depositi epatici di ferro con la tecnica della
susceptometria magnetica, che permette valutazioni addirittura
migliori del dosaggio del ferro nella biopsia. Ma per ora questa
tecnica viene eseguitaselo in 4
centri in
tutto il mondo.
Quadri | Tratto
talassemico | Talassemia
intermedia | Talassemia
major | Talassemia
emoglobina E |
Genetica | Una
mutazione su un gene della β-globina | I due
geni della β-globina presentano la mutazione della
talassemia, in almeno uno dei quali  lieve; una mutazione della
β-globina in combinazione con un eccesso di geni della α-globina
(piu'rara) | Due
geni di β-globina con una grave mutazione talassemica | Un
gene della β-globina con una mutazione talassemica (lieve o
grave) in combinazione con un gene della β-globina, recante la
mutazione puntiforme dell'emoglobina E |
Manifestazioni
cliniche | Nessuna
anemia o anemia lieve, con variabile microcitosi (MCV di 60 fL o
piÂ), non splenomegalia, non alterazioni ossee | Anemia
da lieve a moderata; relativa indipendenza dalle trasfusioni:
splenomegalia e deformita' ossee; grado variabile di sovraccarico
di ferro, dipendente dalla gravita' dell'anemia e dalla richiesta
di trasfusioni | Grave
anemia, richiedente regolari trasfusioni fin dall'infanzia;
splenomegalia e malattia ossea, dipendente dall'efficacia delle
trasfusioni; grave sovraccarico di ferro | Anemia
lieve o grave; relativa dipendenza dalle trasfusioni;
splenomegalia e deformita' ossee; vario grado di sovraccarico di
ferro, dipendente dalla gravita' dell'anemia e dalla richiesta di
trasfusioni |
Gravita' | Asintomatico | Da
forme asintomatiche a forme gravemente sintomatiche | Richiesta
di cure di sostegno per tutta la vita | Da
forme asintomatiche a forme gravemente sintomatiche |
Miglioramento
dei fattori genetici | Presenza
anche di α-talassemia | Presenza
anche di α-talassemia; elevata quantita' di HbF. | Presenza
anche di α-talassemia; elevazione della HbF | Lieve
mutazione β-talassemica; insieme ad α-talassemia; elevazione
della HbF |
Aggravamento
dei fattori genetici | Eccesso
di geni della α-globina | Eccesso
di geni di α-globina (5 o piu') | Non
conosciuto | Mutazione
grave β-talassemica |
Purtroppo
la valutazione dei depositi cardiaci di ferro non può essere
fatta solo sulla base della concentrazione di ferro nel fegato, dei
livelli di ferritina o di ambedue. La valutazione dei depositi di
ferro nel cuore è particolarmente difficile: è in
studio una modificazione della risonanza magnetica.
A parte i
danni legati ai depositi di ferro nei tessuti, il ferro stesso è
altamente tossico perché non è legato alla
transferrina, perché è stata superata la sua capacità
legante. Il ferro libero catalizza la formazione di composti
dell'ossigeno. Una frazione labile di ferro nel plasma può
essere misurata direttamente e fornire utili indicazioni per il
trattamento.
La
terapia chelante raddoppia le aspettative di vita dei pazienti con
talassemia major. La desferoxamina è il chelante più
usato; tuttavia essa ha diversi inconvenienti:
- Necessita della via parenterale (dolorosa e perciò con bassa compliance)
- Presenta effetti collaterali
- È costosa
IlDeferiprone, un chelante da somministrare per bocca, dopo
un'iniziale indecisione, è oggi considerato sicuro ed
efficace. Mentre, come effetti collaterali, non ha conseguenze sul
fegato, esso determina artralgia, nausea e altri sintomi
gastro-intestinali. Determina inoltre fluttuazione nei livelli degli
enzimi epatici, leucopenia e raramente agranulocitosi e deficienza di
zinco. Il deferiporone d'altra parte ha dei vantaggii sulla
deferoxamina:
- Penetra nella cellula attraverso la membrana
- Chela i composti tossici intracellulari dell'ossigeno
- Aumenta i livelli di emoglobina
- Riduce di conseguenza le richieste di trasfusioneÈ più efficace della deferoxamnina nella rimozione del ferro miocardico.
Nuovi
incoraggianti prospettive si ricavano dall'uso contemporaneo di
deferoxamina e di deferiprone: il ferro intracellulare, chelato dal
deferiprone, passa nel plasma dove è presente un ancora più
forte chelante, la deferoxamina, per cui viene eliminata con le urine
una maggiore quantità di ferro.
Altri
chelanti e altre tecniche di dosaggio del ferro intracellulare sono
in preparazione.
Trattamento | Esito |
Trasfusione
-definizione di limite al di sopra del quale  necessaria una
trasfusione (9-10 g/dL invece di 10-12). -uso di tecnica per
l'allontanamento dei globuli bianchi -ricerca dei virus nel
sangue da trasfondere (HBV, HCV, HIUV, HTLV-1) -impianto di un
dispositivo per l'accesso venoso | -minor
sovraccarico di ferro e di allo-immunizzazione -minori reazioni
febbrili non emolitiche da trasfusione, minor trasmissione di
citomegalovirus, minore allo-immunizzazione -sostanziale
riduzione delle infezioni trasmesse con la trasfusione
-trattamento pi efficace e migliore comfort e accettazione da
parte del paziente |
Chelazione
-ricerca della dose di deferoxamina -sviluppo di chelanti orali e
di terapia combinata di chelazione- | -minori
effetti collaterali (sordita', displasia ossea) -migliori
accettabilita' ed efficacia |
Sostegno
endocrino -somministrazione di ormoni di sostituzione
(sessuali, tiroidei, e della crescita) -somministrazione di
sostanze atte alla fertilitË -somministrazione di inibitori
degli osteoclasti | -miglioramento
nella crescita, sviluppo e maturazione sessuale; prevenzione
dellìosteoporosi -induzione della spermatogenesi;
raggiungimento della gravidanza -miglioramento della osteopenia,
osteoporosi e della qualitË della vita |
Vuoi citare questo contributo?