Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Giugno 2005 - Volume VIII - numero 6
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Un
caso di sindrome di Alagille
Divisione
di Pediatria, Ospedale S.S. Annunziata ASL NA 1.
Indirizzo
per corrispondenza: borrelliaurelia@libero.it
Keywords
Alagille syndrome, Prolonged jaundice, Characteristic facies, Case
report
Summary
Alagille syndrome consists of 5 major features comprising paucity of
interlobular bile ducts, characteristic facies, posterior
embryotoxon, vertebral defects and peripheral lung stenosis.
The
authors described the case of a girl who presented, at first clinical
examination, prolonged jaundice and characteristic facies including
broad forehead, deep-set eyes, prominent nose, and pointed chin. The
liver biopsy performed at three months of age showed the
characteristic paucity or interlobular bile ducts. She was treated
with ursodeoxycholic acid. This report also provides a literature
review of Alagille syndrome.
Giunge
alla nostra osservazione per “Ipertransaminasemia” una lattantina
di tre mesi,. terzogenita di una coppia apparentemente sana e non
consanguinea, nata alla 40° settimana da una gravidanza
normocondotta e parto eutocico, con peso alla nascita di 2170 gr
(<5°C), h=47 cm (10 °C), cc=32,5cm (5–10 °C), e
nessun problema alla nascita. Nella anamnesi remota si annovera un
ricovero all'età di 2 giorni, presso la TIN , per “sospetta
sepsi”. In quella occasione, la piccola presentava all' E.O., una
facies insolita con ittero alla cute e alle sclere e un lieve stato
di disidratazione. Il fegato debordava dall'arcata costale di circa
2 cm, era presente un lieve ipertono agli arti superiori con mani
serrate “a pugno”. Le feci erano ipocoliche ad intermittenza, le
urine erano normali. Gli esami ematochimici di routine evidenziarono
un aumento della bilirubina totale 13,2 mg\dl, con bilirubina diretta
di 3,06 mg\dl e delle transaminasi: AST 132; ALT 41; risultarono
nella norma: l'EAB, l'ammoniemia, gli oligotest, il galattosio,
le reazioni sierologiche per la lue (VDRL e TPHA), i Marker
dell'epatite A,B,C, il TORCH, gli anticorpi antivaricella e gli
esami colturali. La piccola nel corso della degenza fu sottoposta a
terapia antibiotica e fototerapia. Venivano inoltre effettuate una
visita oculistica che metteva in evidenza diffuse emorragie
retiniche, una ecocerebrale che mostrava iperecogenicità delle
arterie lenticolostriate, un'ecografia epatobiliare che risultava
nella norma a parte una colecisti contratta ma in sede; un'ecorenale
che risultava nella norma. Al momento del ricovero la piccola
presentava un P= 4,020gr. 5°C, una h= 56 cm (10°C), una cc=38
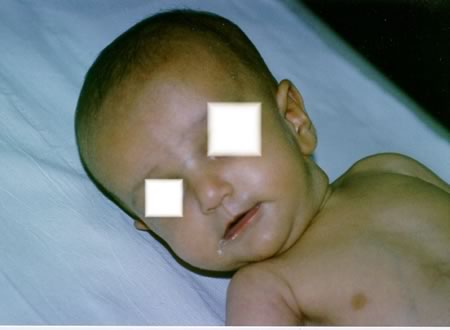
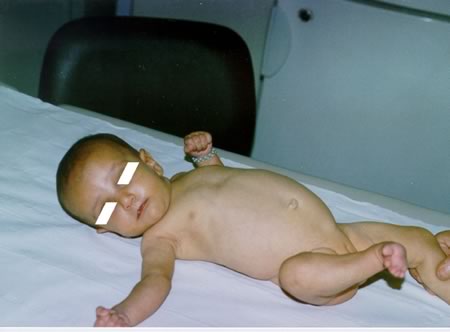
cm (> 10°C) e ittero. All'esame obiettivo sono presenti: una
“facies caratteristica” con fronte prominente, occhi infossati,
ipertelorismo, ponte nasale depresso, mento appuntito, collo corto,
orecchie a impianto basso, F.A. ampia; ittero alla cute e alle sclere
tendente al verdinico (Figure
1, 2), fegato debordante dall'arcata costale di circa 2 cm,
milza nei limiti; cuore: itto in sede, soffio olosistolico 2/6 al
mesocardio, masse muscolari ipotrofiche. Gli esami di laboratorio
eseguiti: immunoglobuline, profilo epatico, marker dell'epatite
A,B,C; profilo coagulativo, V.Wright, TORCH, monotest, alfa1
antitripsina, test del sudore, esami colturali su sangue, urine e
feci hanno confermato l'aumento della bilirubina totale, della
bilirubina diretta, delle transaminasi ( AST= 362, ALT =329) e
mostrato l'aumento della gamma GT=788, della fosfatasi
alcalina=1165, dell'LDH=669 e del colesterolo=389; l'esame delle
urine ha mostrato la presenza di pigmenti biliari e urobilina. In
aggiunta vengono effettuate: una visita oculistica ed un fondo
oculare, un'ecografia epatobiliare, che danno gli stessi risultati
di quelle precedenti. Viene inoltre eseguita una RX del rachide in
toto che risulta nella norma e una visita cardiologica con Ecografia
che evidenzia:
1)
Stenosi del ramo sinistro della polmonare
2)
Persistenza della vena cava superiore sinistra
3) Foro
ovale pervio
Viene
pertanto praticata una biopsia epatica che evidenzia una notevole
paucità dei dotti biliari interlobulari. Per paucità
duttulare si intende l'assenza dei dotti biliari in più del
50% degli spazi portali, quando almeno 10 di essi sono stati
esaminati.
La
piccola viene posta in terapia con acido ursodesossicolico alla dose
di 20- mg\ kg\ die suddivisi in due somministrazioni e a terapia
dietetica con Pregestimil latte.
Nel
nostro caso ci troviamo di fronte ad una patologia caratterizzata da:
1)
Colestasi neonatale intraepatica (con ittero persistente)
2)
Caratteristiche facciali
3)
Anomalie cardiache
4)
Ritardo di crescita
5)
Biopsia epatica suggestiva caratterizzata da paucità congenita
dei dotti biliari interlobulari
Tale
condizione malformativa, a nostro avviso si può far risalire
alla sindrome di Alagille.
Com'è
noto, tale affezione non è altro che una colestasi
intraepatica. “Per colestasi” si intende una ridotta formazione
ed escrezione della bile dovuta a lesione dei dotti biliari intra o
extraepatici, o a un danno degli epatociti. Dobbiamo ricordare che il
fegato del neonato è particolarmente esposto alla colestasi
per l'immaturità delle sue funzioni escretrici e per la
notevole suscettibilità agli insulti virali, batterici o
metabolici. Tale colestasi va quindi sospettata in ogni neonato se
l'ittero non scompare entro la 2° settimana di vita, se è
un ittero a bilirubina coniugata o diretta, se le feci sono acoliche
o ipocoliche, se le urine sono scure. La sindrome di Alagille o
displasia arterioepatica è una malattia autosomica dominante a
penetranza ed espressività variabile spesso si presenta
sporadicamente, in alcuni casi è stata descritta una
familiarità. Il gene coinvolto è denominato JAG1,
l'incidenza è di 1 su 40000.
Sono
stati individuati 5 segni maggiori nella forma completa della
sindrome:
1)
Colestasi intraepatica
2) Facies
particolare (con fronte prominente, occhi infossati, ipertelorismo,
ponte nasale piatto, naso dritto e sottile, mento appuntito, collo
corto, orecchie a impianto basso, profilo del volto piatto).
3)
Difetti cardiaci congeniti (la lesione cardiaca è comunemente
una stenosi polmonare periferica, ma sono state riportate: tetralogia
di Fallot, DIA, coartazione aortica che possono rappresentare il
problema principale).
4)
Anomalie vertebrali (vertebre a farfalla per permanente fusione
dell'arco vertebrale anteriore, fusione di costole) a carico delle
estremità: falangi distali accorciate, inoltre, osteoporosi e
rachitismo resistente alla vit D.
5)
Embriotoxon oculare posteriore che rappresenta un segno patognomonico
presente nel 15% dei casi. Si tratta di un'affezione congenita
dell'occhio nella quale il margine della cornea è opaco.
L'esame con la lampada a fessura mostra inspessimento della linea
di Schwalbe. Si possono inoltre avere strabismo e complicanze a
carico della retina: atrofia, corioretinite. Tali affezioni
potrebbero essere il risultato di una insufficienza di vitamine
liposolubili consecutiva alla malattia epatobiliare.
Anomalie
associate con minore frequenza sono: ritardo di crescita,
interessamento renale (nefropatia tubulare o glomerulare) nel 10% dei
casi, gradi diversi di ritardo psicomotorio e sessuale,
splenomegalia, sordità, voce high-pitched (voce acuta).
L'
associazione tra S. di Alagille e displasia caudale e S. di Alagille
e protoporfirina eritropoietica sono state riportate. E' stata
inoltre osservata una notevole variabilità nella gravità
clinica tra i pazienti affetti. Il 10-15% dei pazienti con sindrome
di Alagille presenta cirrosi. La severità della colestasi dopo
il periodo neonatale è variabile. L'ittero può
sparire o restare lieve ed essere compatibile con una vita quasi
normale. Spesso migliora durante la 2° decade di vita. L'uso
dell'acido ursodesossicolico può migliorare i sintomi e
permettere di rimandare il trapianto ad un'età in cui
eseguirlo presenta minore difficoltà e la terapia successiva
al trapianto comportare un numero minore di effetti collaterali
negativi. Complicanze descritte con maggiore frequenza nell'adulto
sono: insufficienza epatica, pancreatite, carcinoma epatocellulare;
quest'ultima complicanza sarebbe legata alla persistente colestasi.
Nei casi
più gravi, l'ittero persiste e peggiora, il prurito è
incontrollabile, la crescita è severamente compromessa, le
articolazioni ricoperte da xantomi, e la qualità di vita è
pessima. In questi casi l'unica terapia è il trapianto. In
letteratura il 20-25% dei pazienti con S. di Alagille è
candidato al trapianto epatico, tali dati sono sovrastimati poiché,
forniti dagli stessi centri di trapianto.
Nel 1°
anno di vita la facies dei pazienti con S. di Alagille potrebbe non
essere distintiva, ma se sono presenti le caratteristiche facciali
menzionate, la disfunzione epatica e, in particolare l'ipoplasia
dei dotti biliari (come nella nostra paziente), la diagnosi può
essere fatta precocemente. Secondo Sokol e altri le caratteristiche
facciali presenti in pazienti con S. di Alagille potrebbero non
essere caratteristiche dismorfiche primarie ma secondarie alla
presenza della colestasi. La presenza di caratteristiche facciali
tipiche in pazienti con lieve malattia epatica e gli studi dei
parenti di pazienti con S. di Alagille che hanno caratteristiche
facciali tipiche come unici segni clinici sono contro questa ipotesi.
E' stata anche descritta una S. di Alagille senza coinvolgimento
epatico ma con le tipiche caratteristiche facciali.
E'
stato riportato che la S. di Alagille è dovuta a delezione del
braccio corto del cromosoma 20. Alcuni Autori hanno suggerito che la
variabilità nelle manifestazioni fenotipiche potrebbe essere
dovuta ad una delezione di materiale cromosomico di varia entità
tra i differenti casi. Alcuni casi riportati presentano delezione
cromosomica del 20 con punto critico sulla regione p11.2; altri casi
presentano delezione cromosomica del 20 col punto critico che
interessa la regione p11.22 p12.2 o la regione p11.23 e p12.1 o la
regione p 11.13 p 11.2 . Nella nostra paziente e nei suoi familiari
sono stati avviati studi genetici molecolari ma a tutt'oggi non si
hanno ancora risultati. E' stato ipotizzato che la S. di Alagille
costituisca un altro esempio di sindromi genetiche contigue.
L'incidenza della delezione del 20 p nella sindrome di Alagille è
così grande che lo spettro fenotipico di quelle senza
delezioni visibili non è ancora conosciuto. Studi genetici
molecolari di pazienti con una delezione con o senza spettro della S.
di Alagille, e analisi di linkage in casi familiari caratterizzeranno
senz'altro meglio il difetto fondamentale. In opposizione a quanto
detto alcuni Autori ipotizzano che la Sindrome di Alagille sarebbe
causata da un difetto nel metabolismo o nel trasporto di acidi
biliari nelle cellule epatiche o canalicoli biliari. L'associazione
tra S. di Alagille e displasia caudale non può essere
considerata come evento casuale ma come un'estesa alterazione che
interesserebbe il mesoderma assiale infatti alcune caratteristiche
della S. di Alagille sembrano essere manifestazioni di un difetto
mesodermale in quanto cuore e colonna vertebrale si sviluppano dal
mesoderma analogamente alle anomalie presenti nella displasia
caudale. L'associazione inoltre documenta la variabilità
nell'espressione di questa condizione. La S. di Alagille è
stata descritta anche associata alla protoporfiria eritropoietica.
L'analisi
cromosomica ha mostrato anche in questo caso una delezione
cromosomica con punto critico che interessa la regione p 11.23 p
12.2.
La
presenza di carcinoma epatocellulare nei bambini con S. di Alagille e
in un paziente adulto con tale sindrome ma senza cirrosi è
stato messo in relazione alla colestasi cronica ma una spiegazione
alternativa potrebbe essere la perdita di eterozigosi per un gene. Il
gene per la sindrome di Alagille risiederebbe sul braccio corto del
cromosoma 20.
Il
contributo di questo caso clinico è di due ordini:
1) far
conoscere una sindrome rara quale quella di Alagille che è
collocata nel gruppo delle sindromi con fisionomia sui generis,
ritardo di crescita, cardiopatia (displasia caudale, protoporfiria
eritropoietica)
2)
contribuire con la presentazione di questo caso ad un migliore
inquadramento della sindrome.
1.
Mueller RF. The Alagille sindrome (arteriohepatic dysplasia) J Med
Genet. 1987;24(10):621-6. 2. Legius E, Fryns JP, Eyskens B, Eggermout
E, Desmet V, etc. Alagille Syndrome (arteriohepatic dysplasia) and
del (20) (p11.2) Am J Med Genet. 1990;35(4):532-5.
3. Anad
F, Burn J, Matthews D, Cross I, Davison BCC, Mueller R, Sands M,
Lillington DM, Eastham E. Alagille Syndrome and deletion of 20 p. J
Med Genet 1990;27:729-737
4. Watson
GH, Miller V Arteriohepatic dysplasia Familial pulmonary arterial
stenosis with neonatal liver disease. Arch Dis Child 1973;48:459-66
5.
Alagille D, M. Odièvre, M. Gautier, J.P.Dommergues. Hepatic
ductural hypoplasia associated with characteristic facies, vertebral
malformations, retarded physical, mental, and sexual development, and
cardiac murmur. J Pediatr 1975;86:63-71
6
Alagille syndrome associated with caudal dysplasia sequence.
6.
Rodriguez JI, Rivera T, Palacios J. Alagille Syndrome associated with
caudal dysplasia sequenze. Am J Med Genet 1991;40:61-64
7. Teebi
AS, Krishna Murthy DS, Ismail EAR, Redha AA. Alagille Syndrome with
de novo del (20) (p11.2). Am J Med Genet 1992;42:35-38
8)
Henriksen NT, Langmark F, Sorland SJ, Fausa O, Landaas S, Aagenaes O.
Hereditary cholestasis combined with peripheral pulmonary stenosis
and other anomalies. Acta Pediatr Scand 1977;66:7-15
9)
Kocoshis SA, Cottrill CM, O'Connor WN, Haugh R, Johnson GL, Noonan
JA. Congenital heart disease, butterfly vertebrae, and extrahepatic
biliary atresia: A variant of arteriohepatic dysplasia? J Pediatr.
1981;99(3):436-9.
Vuoi citare questo contributo?