
Il
Neuroblastoma (parte prima)
Membro
della Commissione Nazionale Vaccini
Indirizzo
per corrispondenza: bartolozzi@unifi.it
Il
neuroblastoma rappresenta ancor oggi uno dei tumori più
misteriosi: ne sono prova l'estrema eterogeneicità del
quadro clinico e il ventaglio della prognosi che va dalla relativa
benignità dello stadio 4S all'estrema gravità dello
stadio 4, che comporta tutt'oggi una letalità del 40%
nonostante l'uso di farmaci potenti. L'interesse dei ricercatori
è documentato dal numero di pubblicazioni sull'argomento
nel corso degli anni: alla voce NEUROBLASTOMA su PubMed nell'arco
di 55 anni ho trovato 24.757 studi, con un massimo di 1.300 nel
2006.Una recentissima revisione, comparsa sul fascicolo del 23
giugno 2007 del Lancet, offre la possibilità di rivedere i
vari aspetti di questo tumore (Maris JM, Hogarthy MD, Bagatell R,
Cohn SL. Neuroblastoma. Lancet 2007, 369:2106-20).
Il
neuroblastoma è responsabile di più del 7% dei tumori
maligni in soggetti in età inferiore ai 15 anni e di circa il
15% di tutte le morti oncologiche in pediatria. E' questo il più
comune tumore extracranico in pediatria e il tumore più spesso
diagnosticato nell'infanzia.
Negli
anni 80, quando si rese possibile la dimostrazione del tumore
attraverso la ricerca delle catecolamine nelle urine, vennero
intrapresi dei programmi di screening: uno studio giapponese,
eseguito sulle urine raccolte nei bambini a 6 mesi di età,
sembrò dimostrare che una diagnosi precoce poteva influire
l'evoluzione della malattia. Ma due prove cliniche prospettiche
negli Stati Uniti e in Germania hanno dimostrato successivamente che
lo screening non riduce la letalità (Schilling FH et al.,
2002; Woods WG et al, 2002), per cui esso è stato abbandonato.
Aspetti
clinici
Il
neuroblastoma è una malattia della linea simpatico-surrenale
della cresta neurale, per cui i tumori si possono sviluppare ovunque
si trovi il sistema nevoso simpatico. La maggior parte dei tumori
primitivi ha sede nell'addome, metà dei quali a carico della
midollare surrenale. Altre sedi comuni della malattia comprendono:
- Il collo
- Il torace
- La pelvi.
I segni e
i sintomi di presentazione sono fortemente variabili e dipendono in
parte dalla sede del tumore primitivo, ma anche dalla presenza o meno
di metastasi o di sindromi paraneoplastiche. Sebbene vi possano
essere delle sovrapposizioni, si possono presentare tre possibili
scenari:
- Tumori localizzati. Circa il 40% dei pazienti si presenta con malattia localizzata, che può andare dalla scoperta casuale di una massa surrenalica all'ultrasonografia prenatale a tumori molto estesi e localmente invasivi lungo la catena del simpatico nelle età successive. Anche i tumori toracici primitivi possono essere trovati incidentalmente in una radiografia del torace, mentre masse cervicali si possono associare alla sindrome di Horner (ptosi della palpebra superiore + miosi + restringimento della fessura palpebrale). Tumori paraspinali del torace, dell'addome e della pelvi si ritrovano nel 5-15% dei pazienti: questi tumori possono estendersi ai forami neurali intervertebrali (neuroblastomi a clessidra), determinando sintomi da compressione delle radici nervose e del midollo spinale. Circa il 5% di tutti i neuroblastomi sono diagnosticati in pazienti che hanno segni neurologici in rapporto all'interessamento del midollo, con stanchezza, dolore e perdita della sensibilità. I tumori localizzati in generale rispondono al trattamento chemioterapico e/o alla laminectomia: poiché la laminectomia può avere a distanza delle conseguenze, la maggior parte dei ricercatori raccomanda la chemioterapia come intervento di scelta per i neuroblastomi paraspinali sintomatici. In questi pazienti si osservano due importanti sindromi paraneoplastiche: la diarrea acquosa intrattabile, che cessa con l'asportazione del tumore, e la sindrome opsoclono-mioclono, vista nel 2-4% dei pazienti con neuroblastoma: essa consiste in movimenti rapidi degli occhi, atassia e movimenti muscolari irregolari. In questi casi la prognosi è favorevole, ma il 70-80% di questi bambini presentano successivamente deficit neurologici permanenti.
- Malattia metastatica. Circa la metà dei pazienti si presenta con metastasi ematogene. Le metastasi possono essere a distanza, come nella parte corticale delle ossa, nel midollo, nel fegato e nei linfonodi regionali non contigui, o per diffusione del tumore primitivo nei linfonodi adiacenti. A differenza di quanto si osserva nei bambini con tumori localizzati, questi bambini con tumori metastatizzati appaiono profondamente ammalati al momento della diagnosi. Questi tumori hanno una tendenza inspiegabile a metastatizzare nelle ossa dell'orbita e a determinare delle ecchimosi delle orbite e proptosi. Talvolta questi pazienti hanno ipertensione, renina-mediata, dovuta a compromissione della vascolarizzazione renale. Una diffusione al sistema nervoso centrale può essere osservata con levolvere della malattia.
- Malattia 4S. D'Angio e coll. descrissero per primi le caratteristiche di questa forma che si verifica nel 5% dei pazienti (D'Angio GJ et al, 1971). Questi bambini hanno un piccolo tumore primitivo con metastasi al fegato, alla cute e al midollo osseo, che regrediscono quasi sempre spontaneamente. Tuttavia i bambini in età inferiore ai 2 mesi possono avere una diffusione epatica estesa e rapidamente progressiva, che può addirittura compromettere la respirazione.
Diagnosi
La
diagnosi di neuroblastoma si basa sulla presenza aspetti
isto-patologici caratteristici o per la presenza di cellule tumorali
nell'ago aspirato di midollo osseo, accompagnate da aumentate
concentrazioni di catecolamine urinarie.
I
pazienti ad alto rischio hanno spesso:
- Aumentate concentrazioni di lattico-deidrogenasi sierica
- Ferritina
- Cromagranina
Ma
bisogna ricordare che questi aspetti sono non-specifici nella
popolazione in generale, per cui essi non sembrano avere un
significato prognostico alla luce delle moderne co-variate. Gli
indicatori genetici specifici e le valutazioni isto-patologiche sono
fondamentali per la determinazione del trattamento, specialmente nei
bambini in età inferiore ai 18 mesi (figura 1). Le biopsie
tumorali sono sufficienti per le analisi molecolari genetiche, per
cui queste ricerche vanno incoraggiate al momento della diagnosi.
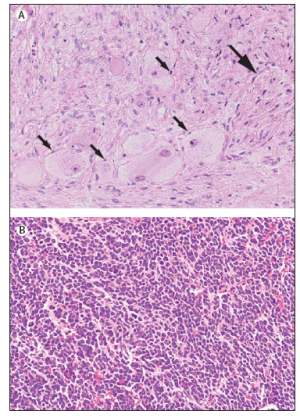
Figura
1. Tumori metastatici con diversi gradi di differenziazione.
(Maris JM et al, 2007).
- le cellule di Schwann e le cellule dei gangli (indicate con le frecce) sono abbondanti nel neuroblastoma ricco di stroma
- nei neuroblastomi poveri di stroma sono evidenti cellule rotondeggianti, stipate, con scarso citoplasma.
Valutazione
clinica della malattia
La
tomografia computerizzata (TC) è il metodo preferito per la
valutazione dei tumori dell'addome, della pelvi e del mediastino.
La
risonanza magnetica (RM) va meglio per le lesioni paraspinali e
diviene essenziale quando si vogliano valutare le estensioni dei
tumori intra-foraminali per studiare l'eventuale compressione del
midollo.
La
scintigrafia con betaiodobenzylguanidina (MIBG) ha un'aumentata
sensibilità e specificità in confronto a precedenti
traccianti. Poiché l'MBIG si concentra selettivamente in più
del 90% dei neuroblastoma, scintigrafia con MBIG è un metodo
altamente specifico per la valutazione del tumore primitivo e per la
malattia metastatica (figura 2).
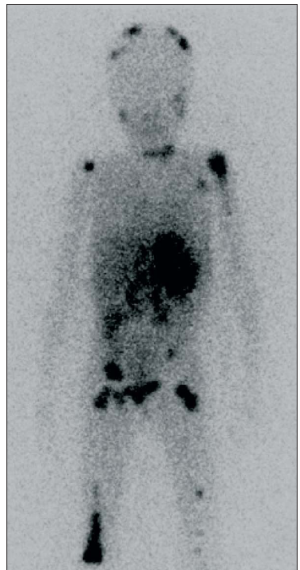
Figura
2. La betaiodobenzylguanidina (MIBG) mette in evidenza le sedi
multiple della malattia, incluso il tessuto osseo e il tessuto
sottocutaneo. (Maris, JM, 2007)
Poiché
un certo numero di neuroblastoma altamente non differenziati possono
non concentrare MBIG, il TC-difosfonato può essere usato nelle
ricerche sull'osso, quando la MBIG sia risultata negativa o non sia
disponibile.
La MBIG
Viene anche raccomandata per la rivalutazione durante e dopo la
terapia nei pazienti ad alto rischio, che erano risultati positivi
all'MBIG alla diagnosi.
La
malattia del midollo osseo va valutata anche con altri mezzi, come
aspirati e biopsie midollari. Vi è un numero elevato di
pubblicazioni sull'uso dell'immunocitochimica e su tecnologia
basate sulla Reazione Polimerasica a Catena (PCR) per mettere in
evidenza le cellule del neuroblastoma, come la tirosino-idrossilasi,
la GD2 sintasi, la PgP9.5 nel midollo e nel sangue alla diagnosi e
alla ricerca della malattia residua minima.
Stratificazione
del rischio
La
valutazione del rischio dipende da molti aspetti clinici e biologici.
Poiché la valutazione del rischio varia fortemente da un Paese
all'altro, negli ultimi due anni l'International Neuroblastoma
Risk Group (INRG) ha rivedute le cartelle cliniche di 11.054 bambini,
trattati in Europa, Giappone, Canada, USA e Australia fra il 1974 e
il 2002. E' stato così ottenuto un consenso:
- Riguardo all'età: prima e dopo i 18 mesi;
- Lo stadio
- Lo stato MYCN
Variabili
cliniche
L'International
Neuroblastoma Staging System ha sviluppato i criteri di stadiazione,
che sono stati accolti nel mondo (vedi Tabella 1). Questi studi hanno
dimostrato che le caratteristiche radiologiche del tumore primario
sono utili per predire il trattamento chirurgico migliore e più
di successo. Gli aspetti loco radiologici sono stati usati per
distinguere i tumori locoregionali che non interessano strutture
locali (INRG stadio L1) dai tumori localmente invasivi (INRG stadio
L2). Gli stadi M ed MS sono stati proposti per categorizzare tumori
che siamo ampiamente metastatici o che abbiano un comportamento 4S.
Per
quanto riguarda l'età è preferibile tener conto
dell'età da 15 a 18 mesi, invece di quella a 12 mesi.
Tabella
1. Stadiazione INSS
Stadio |
Caratteristiche
del tumore |
1 |
Tumore
localizzato con completa escissione grossolana, con o senza
malattia microscopiva residua, rappresentata da linfonodi
ipsolaterali negativi microscopicamente per tumore (i linfonodi
aderenti al tumore e rimossi con il tumore primitivo possono
essere positivi). |
2A |
Tumore
localizzato con escissione grossolana incompleta, con linfonodi
ipsolaterali non aderenti, positivi per tumore. |
2B |
Tumore
localizzato con o senza escissione completa grossolana, con
linfonodi ipsolaterali non aderenti positivi per tumore. I
linfonodi controlaterali ingranditi debbono essere negativi
microscopicamente. |
3 |
Tumore
unilaterale non resecabile con o senza ineteressamento dei
linfonodi regionali o tumore localizzato unilaterale con
interessamento dei linfonodi regionali contro laterali o tumore
della linea mediana con estensione bilaterale dell'infiltrazione
(non resecabile) o con interessamento dei linfonodi |
4
|
Ogni
tumore primario con disseminazione a linfonodi distanti, all'osso,
al midollo osseo o ad altri organi (eccetto lo stadio 4S), |
4S |
Tumore
primario localizzato in lattanti più giovani di un anno
(come definiti dagli stadi 1, 2A e 2B), con disseminazione
limitata alla pelle, al fegato o al midollo osseo (< 10% di
cellule maligne) |
Stadio |
Età |
MYCN |
Ploidia |
Istologia |
Altro |
Rischio |
1 |
Basso | |||||
2A/2B |
Non
amplificato
Non
amplificato
Non
amplificato
Amplificato |
>50%
di resezione
<50%
di resezione
Soltanto
biopsia |
Basso
Intermedio
Intermedio
Alto | |||
3 |
<
547 giorni
≥ 547
giorni
≥ 547
giorni |
Non
amplificato
Non
amplificato
Amplificato
Non
amplificato |
Favorevole
Sfavorevole |
Intermedio
Intermedio
Alto
Alto | ||
4 |
<
365 giorni
<365
giorni
365-547
giorni
365-547
giorni
365-547
giorni
365-547
giorni
≥ 547
giorni |
Amplificato
Non
amplificato
Amplificato
Non
amplificato
|
DI
= 1
DI>1 |
Sfavorevole
Favorevole |
Alto
Intermedio
Alto
Alto
Alto
Intermedio
Alto | |
4S |
<365
giorni
<365
giorni
<
365 giorni
<365
giorni
<365
giorni
<365
giorni |
Non
amplificato
Non
amplificato
Perduto
Non
amplificato
Non
amplificato
Amplificato |
DI>1
DI=1
Perduto |
Favorevole
Perduto
Sfavorevole |
Asintomatico
Sintomatico |
Basso
Intermedio
Intermedio
Intermedio
Intermedio
Alto |
Istologia
del tumore
Nel 1984
Shimada e coll (J Natl Cancer Inst, 1984, 73:415-17) presentarono
uno schema classificativo mettendo in relazione gli aspetti
isto-patologici del tumore con il comportamento clinico. I tumori
venivano classificati come favorevoli e non favorevoli a seconda del
grado di differenziazione neuroblastica, il contenuto di stroma
Schwanniano, l'indice di mitosi carioressi e l'età alla
diagnosi. Lo schema è stato successivamente modificato.
Variabili
biologiche
L'aberrazione
genetica più spesso associata a prognosi cattiva nel
neuroblastoma è l'amplificazione genomica di MYCN. Questa
amplificazione avviene in circa il 20% dei tumori primitivi ed è
fortemente correlata con lo stadio avanzato della malattia e con
l'insufficienza del trattamento.
La
delezione del braccio corto del cromosoma 1 (1p) può essere
trovata nel 25-35% dei neuroblastoma. Questa delezione non solo si
correla con l'amplificazione MYCN ma anche con lo stadio avanzato
della malattia.
La
perdita allelica di 11q è presente nel 35-45% dei tumori
primitivi. Questa aberrazione genomica si vede raramente in tumori
con amplificazione MYCN, anche se rimane fortemente associata con
aspetti ad alto rischio.
Un
guadagno di 1-3 copie 17q, spesso attraverso traslocazioni non
bilanciate con il cromosoma 1 o 11, si possono anche correlare con un
fenotipo più aggressivo.
L'indice
DNA è anche un indicatore prognostico per pazienti più
giovani di due anni, che hanno malattia disseminata. Il contenuto in
DNA del neuroblastoma riguarda due ampie categorie: quasi diploidi e
iperdiploidi (spesso triploidi). Modelli genetici di neuroblastoma
suggeriscono che i tumori meno aggressivi hanno un fondamentale
difetto nella mitosi, che può spiegare l'impressione che la
prognosi dei quasi tetraploidi sia più favorevole. Al
contrario i neuroblastomi più maligni hanno un difetto
fondamentale nella stabilità genetica, risultante in un
riarrangiamento cromosomico, in traslocazione non bilanciata e nel
mantenimento di un contenuto quasi diploide.
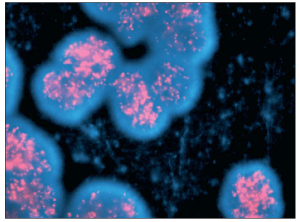
Figura
3. Amplificazione MYCN dimostrata dalla fluorescenza nella
idridizzazione in situ (FISH) (Maris, JM, 2007)
La
presenza di copie multiple di MYCN può essere osservata nelle
cellule tumorali usando il probe marcato MYCN.
Vuoi citare questo contributo?
G. Bartolozzi. IL NEUROBLASTOMA (PARTE PRIMA).
Medico e Bambino pagine elettroniche 2007; 10(7)
https://www.medicoebambino.com/_tumori_neuroblastoma_linfonodi_midollo_osseo