Settembre 2014 - Volume XVII - numero 7
M&B Pagine Elettroniche
I Poster degli specializzandi
Porfiria acuta intermittente: un caso "eccezionale"
1Scuola di Specializzazione di Pediatria, Università di Modena e Reggio Emilia
2Dipartimento Materno-Infantile, Azienda Ospedaliero-Universitaria Policlinico di Modena
3UO Medicina Interna 2, Ambulatorio delle Porfirie e dei Disturbi del Metabolismo degli Aminoacidi; Azienda Ospedaliero-Universitaria Policlinico di Modena, Dipartimento di Scienze Medico-Chirurgiche Materno-Infantili e dellAdulto, Università di Modena e Reggio Emilia 4Servizio di Neuropsichiatria Infantile, Arcispedale S. Maria Nuova, Reggio Emilia
2Dipartimento Materno-Infantile, Azienda Ospedaliero-Universitaria Policlinico di Modena
3UO Medicina Interna 2, Ambulatorio delle Porfirie e dei Disturbi del Metabolismo degli Aminoacidi; Azienda Ospedaliero-Universitaria Policlinico di Modena, Dipartimento di Scienze Medico-Chirurgiche Materno-Infantili e dellAdulto, Università di Modena e Reggio Emilia 4Servizio di Neuropsichiatria Infantile, Arcispedale S. Maria Nuova, Reggio Emilia
Indirizzo
per corrispondenza:
chiarasandrin@hotmail.it
Le porfirie, disordini metabolici ereditari ed eterogenei, comprendono 8 quadri clinici causati da mutazioni enzimatiche nella catena biosintetica delleme che esitano in un accumulo di precursori tossici e patogenetici (Figura 1).
Si distinguono porfirie acute e porfirie cutanee. Le prime, tra le quali la più comune è la porfiria acuta intermittente (AIP), si manifestano con crisi neuroviscerali e a carico del SNP, anche letali, descritte in letteratura quasi esclusivamente in soggetti puberi.
A.Z., 6 anni, figlio di cugini di 1° grado, veniva valutato per rifiuto dellalimentazione, addominalgia ricorrente da due mesi che da oltre 2 settimane si associava a difficoltà alla deambulazione. Obiettivamente: marcata oppositività comportamentale, stato di ipotrofia muscolare e iponutrizione e un'andatura steppante con ipostenia severa alla muscolatura distale delle estremità.
Le indagini ematochimiche, neuroradiologiche, infettivologiche (compresa rachicentesi con esame del liquor) e autoimmunitarie risultavano tutte negative, salvo per un quadro elettromiografico di neuropatia assonale cronica a carico del nervo peroneo e mediano bilateralmente.
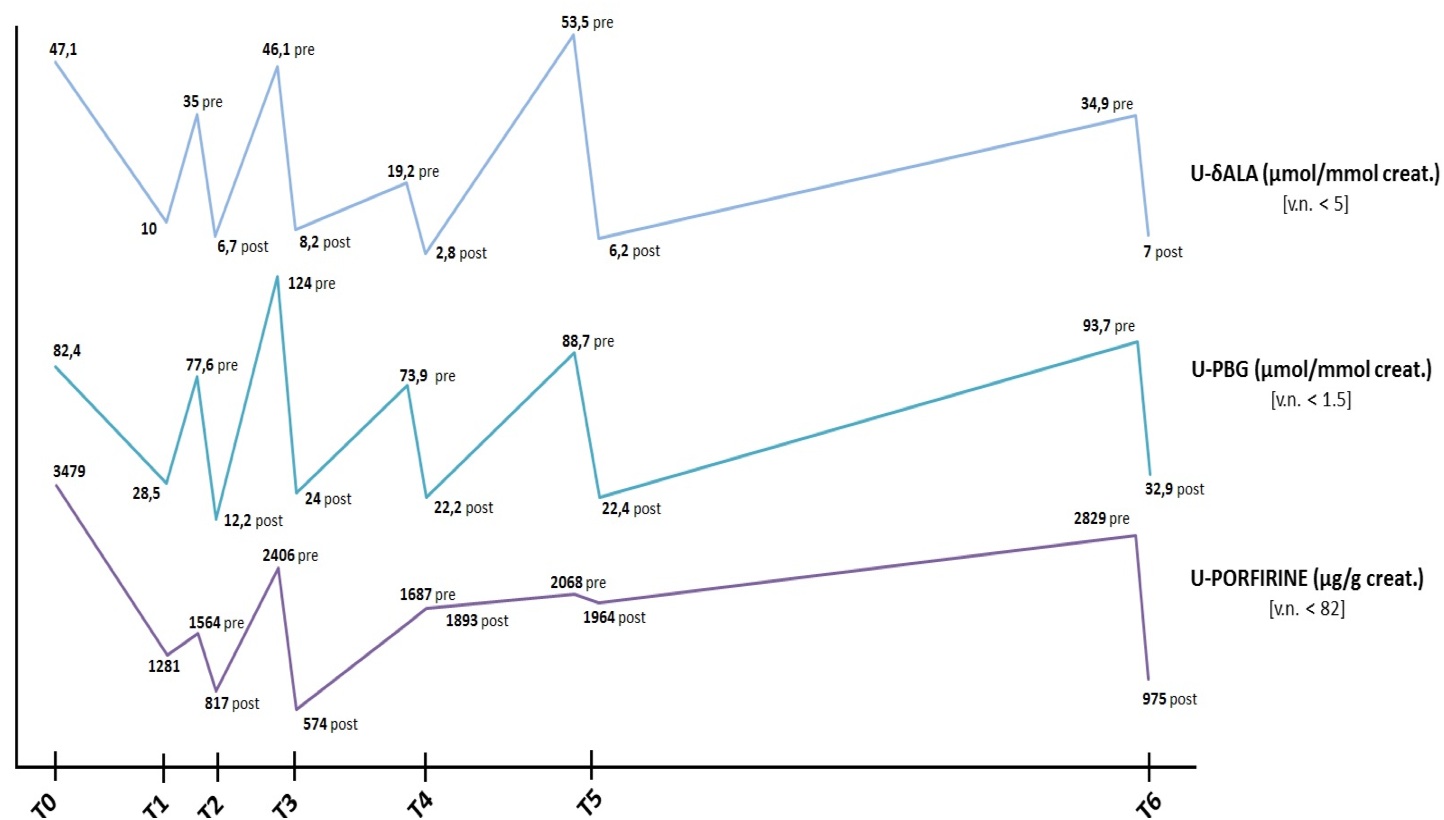
Per tale riscontro si eseguiva anche una valutazione delle porfirine urinarie totali, con esito positivo a valori particolarmente elevati. L'analisi delle frazioni porfiriniche era suggestiva per AIP, in seguito confermata dallanalisi genetica. L'infusione di soluzione glucosata al 10% e, a cicli ripetuti, di emina umana (3 mg/kg) ha portato a un radicale miglioramento, che persiste attualmente, sia dei livelli porfirinici nelle urine sia della sintomatologia addominale e neuro-psichiatrica.
I sintomi più frequenti dellAIP sono addominalgia, nausea, vomito e tachicardia. Tra i sintomi neurologici la neuropatia coinvolge il motoneurone con ipostenia a carico della muscolatura prossimale degli arti. La diagnosi di AIP è decisamente inusuale in un paziente di 6 anni; lesordio clinico, nonostante la presenza di addominalgia, appare relativamente atipico anche dal punto di vista neurologico. L'eccezionalità" di questo caso, per letà dell'esordio e per le caratteristiche cliniche intrinseche, sottolinea come la diagnosi di porfiria acuta non debba e non possa essere esclusa in età pediatrica.
Figura
1.
A.Z., 6 anni, figlio di cugini di 1° grado, veniva valutato per rifiuto dellalimentazione, addominalgia ricorrente da due mesi che da oltre 2 settimane si associava a difficoltà alla deambulazione. Obiettivamente: marcata oppositività comportamentale, stato di ipotrofia muscolare e iponutrizione e un'andatura steppante con ipostenia severa alla muscolatura distale delle estremità.
Le indagini ematochimiche, neuroradiologiche, infettivologiche (compresa rachicentesi con esame del liquor) e autoimmunitarie risultavano tutte negative, salvo per un quadro elettromiografico di neuropatia assonale cronica a carico del nervo peroneo e mediano bilateralmente.
Per tale riscontro si eseguiva anche una valutazione delle porfirine urinarie totali, con esito positivo a valori particolarmente elevati. L'analisi delle frazioni porfiriniche era suggestiva per AIP, in seguito confermata dallanalisi genetica. L'infusione di soluzione glucosata al 10% e, a cicli ripetuti, di emina umana (3 mg/kg) ha portato a un radicale miglioramento, che persiste attualmente, sia dei livelli porfirinici nelle urine sia della sintomatologia addominale e neuro-psichiatrica.
I sintomi più frequenti dellAIP sono addominalgia, nausea, vomito e tachicardia. Tra i sintomi neurologici la neuropatia coinvolge il motoneurone con ipostenia a carico della muscolatura prossimale degli arti. La diagnosi di AIP è decisamente inusuale in un paziente di 6 anni; lesordio clinico, nonostante la presenza di addominalgia, appare relativamente atipico anche dal punto di vista neurologico. L'eccezionalità" di questo caso, per letà dell'esordio e per le caratteristiche cliniche intrinseche, sottolinea come la diagnosi di porfiria acuta non debba e non possa essere esclusa in età pediatrica.
Vuoi citare questo contributo?