Congenital
pulmonary airway malformations (CPAM) include a wide spectrum of
developmental anomalies. Almost 50% of the cases are diagnosed in
utero. These appear as hyperechoic, cystic or mixed lesions.
Prenatal clinical course varies from serious complications in
utero to polyhydramnios with premature labor. Hydrops and fetal
demise is reported in as many as 25-30% of cases. Postnatal
presentation ranges from severe respiratory distress to symptoms
related to pulmonary hypoplasia, left to right shunting in
sequestration or overinflation and pneumothorax. When no prenatal
diagnosis is made, children may remain asymptomatic or may
present, later in their lives, with complications of recurrent
pneumonias or spontaneous pneumothorax. Surgical resection
represents the standard for symptomatic cases at birth and later
in children with respiratory symptoms. Even though the management
for asymptomatic cases remains controversial, early surgical
excision to avoid recurrent infections, pneumothorax and
malignancy is recommended. New instrumentation and advanced
skills make thoracoscopic lobar lung resection the preferred
approach.
Le
malformazioni congenite polmonari (CPAM) includono un ampio
spettro di lesioni secondarie ad anomalie dello sviluppo del
polmone1. Nel 50% dei casi la diagnosi avviene in utero
con lidentificazione dei 3 aspetti morfologici ecografici
tipici: le macro e micro cisti (CPAM tipo I e II) e le forme solide2
Tipo III e PS (sequestro polmonare).
La storia
naturale della malformazione varia dalla comparsa di polidramnios
causa di prematurità, allidrope (con rischio di morte
in utero nel 25-30% dei casi), alla mancata visualizzazione della
lesione a fine gravidanza (15%). Nei casi più gravi vi è
lindicazione al posizionamento di uno shunt toraco-amniotico o
allintervento chirurgico in utero. Nella gran parte dei casi
le lesioni cistiche CCAM Tipo I e II, presentano un incremento delle
dimensioni fino alla 32 settimana di gestazione. Le forme solide,
invece, CCAM tipo III e PS, sono considerate le più stabili:
crescono con il feto e subiscono, a fine gravidanza, un arresto della
crescita. Lenfisema congenito presenta un andamento simile al
tipo III, scompare nel 3° trimestre di gravidanza ed è
visibile al controllo TAC postnatale. Segni ecografici prenatali di
compressione del mediastino e di idrope impongono lespletamento
del parto in un centro di 3° livello.
In tutti
i casi con regressione della lesione in gravidanza, o asintomatici
alla nascita, la malformazione va ricercata nei primi mesi di vita
con TAC del polmone.
I casi
sintomatici alla nascita possono presentare sintomi da ipoplasia
polmonare, distress respiratorio, pneumotorace, shunt
sinistro-destro, tipico del sequestro polmonare. La terapia
chirurgica è la lobectomia. Per i pazienti asintomatici,
invece, lindicazione alla resezione polmonare è
auspicabile entro lanno di vita. Polmoniti ricorrenti e
pneumotorace sono, invece, lespressione clinica della presenza
di una CPAM in pazienti senza diagnosi prenatale. Nel 53% dei casi le
CPAM sono associate a overlapping lesion: enfisema
perilesionale, bronchiectasie, sequestro polmonare. Per questa
ragione oltre il 60% delle malformazioni presentano una discrepanza
tra la diagnosi pre- e post-operatoria. Inoltre, estesi fenomeni
flogistici, coinvolgono le malformazioni e sembrano essere presenti
anche nei pazienti asintomatici sin dalla nascita. In tutti i casi, e
soprattutto per quelli con diagnosi tardiva o manifestazione clinica
di una lesione misconosciuta, la resezione lobare deve avvenire il
più presto possibile per evitare infezioni ricorrenti,
degenerazione maligna e pneumotorace. La crescita polmonare
compensatoria è eccellente fino al 5°-7° anno di età.
Lapproccio
chirurgico toracoscopico mini invasivo è la metodica di
scelta: riduce i tempi di degenza ospedaliera e il fabbisogno di
analgesia post operatoria. Il risultato estetico è eccellente.
Con la lobectomia vengono asportate anche le overlapping
lesion, potenziali sedi di processi pneumonici.
Cosa
sono e come si classificano
Le CCAM
(congenital cystic adenomatoid malformation) sono masse
intrapolmonari caratterizzate da un incremento della componente
adenomatoide dei bronchi terminali. Le cisti che ne derivano
presentano dimensioni variabili da un millimetro a più di 10
centimetri. Sono evidenti sottili connessioni con lalbero
tracheo-brionchiale, non coinvolti negli scambi gassosi3.
Negli
anni 70 fu introdotta da Stocker la classificazione in tre tipi,
basata sulla grandezza della lesione. Nel 2001 la denominazione delle
CCAM fu modificata in CPAM: Congenital pulmonary airway
malformation per includere cinque tipi di lesione e le
relative origini della malformazione, dalla trachea, dai bronchi, dai
bronchioli, alveoli, e acini. Questa nuova suddivisione, pur di
scarsa applicazione in diagnosi prenatale, è molto utile per
la definizione anatomopatologica e anche perché tiene conto
delle varie possibili coesistenze malformative come lenfisema
lobare, (PIPE) e il sequestro intralobare (ILE)4,5.
Per
comodità di definizione diagnostica, lecografia
prenatale mantiene la suddivisione delle lesioni in cistiche (CCAM 1
> 2 cm, CCAM 2 < 1 cm), e solide (CCAM 3 con microcisti). Più
semplicemente, dal punto di vista clinico, si ricorre alla
suddivisione ecografica, proposta da Adzick, in due categorie:
macrocisti, con cisti sopra i cinque millimetri, e microcisti con
aspetto di massa solida.
Il
sequestro polmonare non ha comunicazione con lalbero
tracheo-bronchiale ma presenta uno o più rami arteriosi a
diretta dipendenza dallaorta toracica. La grande maggioranza
delle forme intralobari è localizzata ai lobi inferiori,
mentre le extralobari sono generalmente reperite nella regione
toracica postero mediale, talvolta sovra o infradiaframamtica. Il
sequestro polmonare è spesso associato alle CCAM, soprattutto
di tipo II.
Lenfisema
lobare congenito è una malformazione che sembra originare da
unostruzione bronchiale parziale che induce un effetto a tipo
valvola. Locclusione può essere intrinseca da
tracheomalacia o estrinseca (vascolare o da cisti broncogena). Questa
condizione di overinflation polmonare può essere
isolata o molto spesso associata alle CCAM o ai sequestri polmonari.
Diagnosi
pre e post natale e implicazioni nel management
La gran
parte delle CPAM è ora diagnosticabile in gravidanza6.
Laccuratezza della diagnosi prenatale ultrasonografica aumenta
dal 59%, a 32 settimane, all81% a 36 settimane di gestazione.
Il
follow-up prenatale include la sorveglianza della comparsa di segni
di complicanze quali il coinvolgimento della lesione allintero
emitorace, la compressione cardiaca e mediastinica (20% dei casi), la
comparsa di segni di impegno cavale quali il polidramnios e lidrope.
La
presenza di idrope fetale (25-30% delle complicanze prenatali)
delinea un punto di svolta nella malattia. È considerato un
elemento di aggravamento prognostico e costituisce una indicazione al
drenaggio toraco-amniotico o allintervento in utero, a
prevenzione della complicanza estrema e mortale dello stato
anasarcatico (30% delle complicanze)7.
In epoca
neonatale la radiografia del torace può apparire normale anche
in presenza di lesione già confermata in gravidanza. Anche per
questi casi è lindagine radiologica di elezione.
Definisce sede, tipo, estensione della lesione. Lesame con
contrasto è raccomandato per tutti i casi, siano essi già
diagnosticati in utero, asintomatici alla nascita o con storia di
scomparsa della lesione al follow-up prenatale.
Clinica
e trattamento
Quando la
diagnosi prenatale è nota l80% dei casi è
asintomatico alla nascita.
Se la
diagnosi avviene in epoca post natale, o più tardivamente, i
sintomi classici sono:
distress
respiratorio: sia nelle prime settimane di vita, come anche nel
lattante e anche nel bambino più grande. Il sospetto
diagnostico è la presenza di una CCAM.
infezioni
respiratorie ricorrenti nei bambini dopo i due anni di vita. Una
storia di polmonite recidivante sempre nello stesso lobo deve far
pensare alla presenza di un soggiacente sequestro polmonare.
I sintomi
di una CCAM non diagnosticata e non trattata possono variare
dallepisodio di distress respiratorio acuto, alla tosse
cronica, alle infezioni toraciche ricorrenti8. La
lobectomia è la procedura di scelta: evita la ricorrenza di
recidive locali secondarie allelevata incidenza di lesioni
malformative coesistenti, le overlapping lesion. Tra
queste va citato soprattutto lenfisema, molto spesso associato
alla malformazione principale e responsabile, se non asportato in
toto, di flogosi cronica. Lapproccio chirurgico guarda con
attenzione al risparmio del tessuto polmonare in tutte le epoche
dellinfanzia e delladolescenza. Tuttavia, quando il
compenso polmonare è fisiologico, e cioè prima dei 5-7
anni di vita, la metodologia di scelta è la lobectomia. Nel
bambino più grande e nelladolescente può essere
discussa una metodologia di risparmio polmonare sotto forma di
asportazione della lesione a segmenti, a condizione che non vi siano
stati pregressi episodi infiammatori che mal delimitano lestensione
della malformazione iniziale.
Per
questa ragione il trattamento chirurgico precoce va incoraggiato
anche per le malformazioni asintomatiche. Lobiettivo è
quello di evitare il potenziale rischio di infezioni ricorrenti, il
coinvolgimento flogistico di segmenti polmonari attigui sani, la
trasformazione maligna (carcinoma, blastoma pleurico)9,
linsorgenza di scoliosi toracogeniche secondarie a resezioni
polmonari molto ampie.
Suggerimenti
Valutazione
post natale
RX
torace:
in prima
giornata solo per casi con deviazione mediastinica nota o
sintomatici
TAC
torace:
in prima
giornata in caso di distress respiratorio o di compressione
mediastinica nota in prenatale
ai tre
mesi di vita nei pazienti asintomatici, con diagnosi prenatale nota
o con storia di regressione della lesione in gravidanza
Timing
chirurgico
Resezione
toracoscopica o toracotomica:
in
urgenza nel neonato se sintomatico entro lanno di vita nei
casi asintomatici, noti in diagnosi prenatale in tutti gli altri
casi, diagnosticati più tardivamente, a risoluzione della
sintomatologia respiratoria e dei segni di flogosi.
Nella
Tabella uno viene riportato il follow-up di un
gruppo di pazienti noti in diagnosi prenatale sin dalla 23
settimana di gestazione. Alcune considerazioni:
lincidenza
di lesioni associate10-12 documentate allesame
anatomopatologico, è risultata elevata, pari al 53%.
Vi è
una corrispondenza con laspetto ecografico prenatale e quello
perinatale: le compressioni mediastiniche documentate in utero poco
prima della nascita necessitano di assistenza rianimatoria perinatale
in un centro di terzo livello dove sia possibile eseguire anche la
chirurgia nei primissimi giorni di vita.
La
strategia chirurgica dovrebbe considerare lalta incidenza di
lesioni istologiche associate, unitamente allaumentato rischio
di complicanze tardive nel ritardato o mancato trattamento
chirurgico13.
Tabella.
Aspetti clinici e patologici pre- e post natali in un gruppo di 25
pazienti operati per CAM (Burlo 1996-2006)
Diagnosi
Prenatale
Shift
Mediastinico
Parto
Distress
fetale
Peso
alla nascita
Chirurgia
Intervento
Istologia
CAM
1 8 casi
3
Casi
Taglio
cesareo: 32-36 sett
4
casi
3
Casi
1140-2700
g
3
mesi
(3
casi)
Resezione
CAM
1 + Lesioni associate
(4
casi)
at
birth
(5
casi)
Lobectomia
CAM
2
11
casi
6
casi
Taglio
cesareo:
36-38
sett
(6
casi)
6
Casi
2400-2700
g
3
mesi
(5
casi)
Resezione
CAM
2 + Enfisema lobare
(2
casi)*
at
birth
(6
casi)
Lobectomia
CAM
3
6
feti
Parto
vaginale
40
sett
3000
g
5
mesi
(6
casi)
Lobectomia:
3
casi
CAM
2 + ILS
Resezione
-
LS (2 casi)*
ELS
+ CAM 3
(2
casi)
(*)
Casi che hanno presentato recidiva dopo resezione polmonare
CASI
CLINICI
Caso
1
S. è
un bambino di 4 anni in cui era stata fatta diagnosi prenatale di
malformazione basale sinistra alla 23 settimana di gestazione.
A fine gravidanza la lesione non era più visibile per cui,
dopo la nascita non era più stata ricercata. Dalletà
di 2 anni, tuttavia erano comparsi episodi polmonari infettivi
recidivanti sempre alla base inferiore di sinistra.
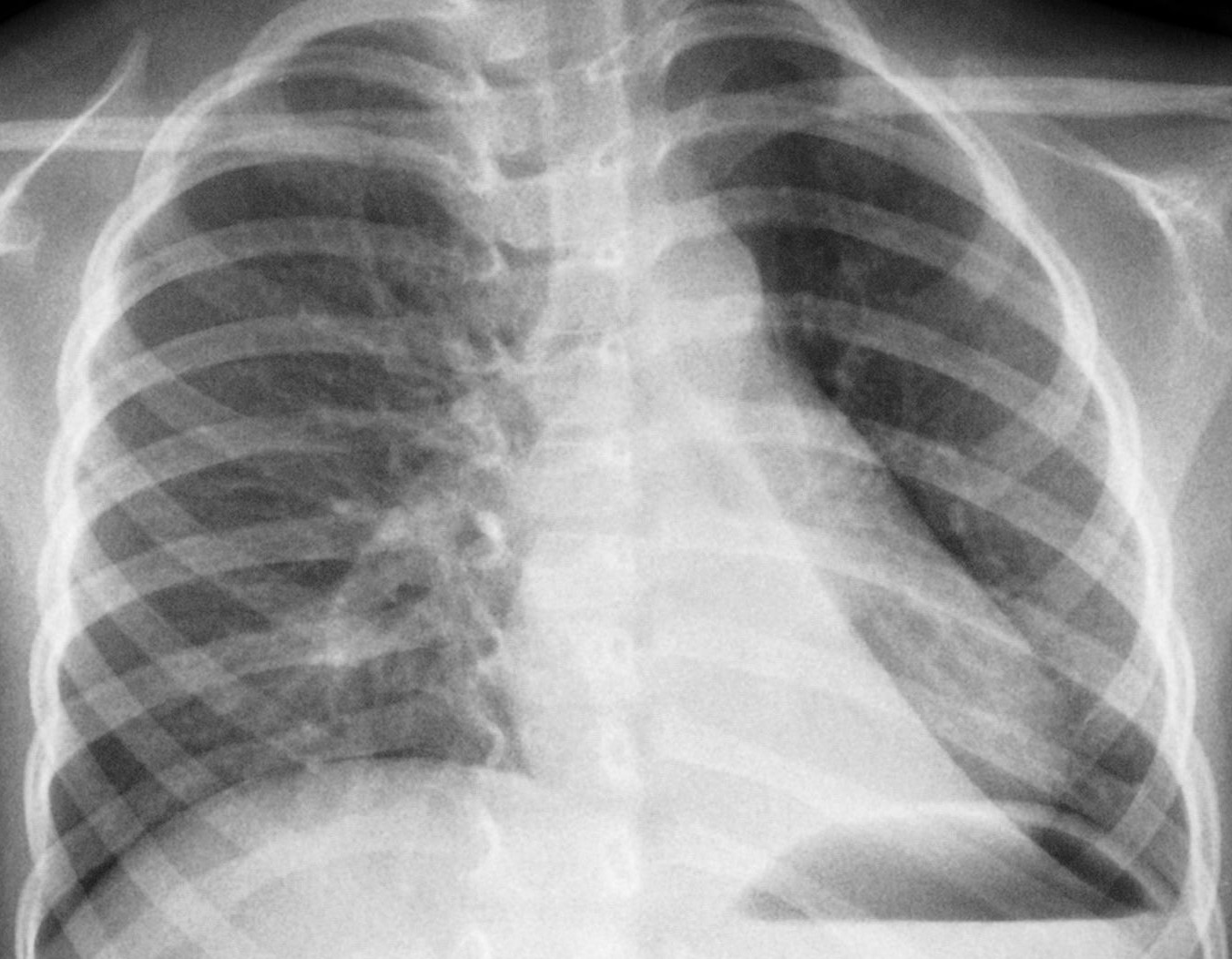
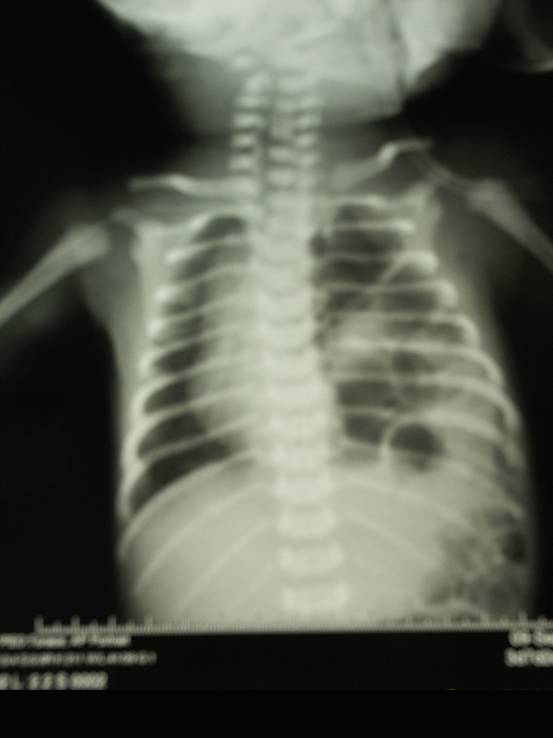
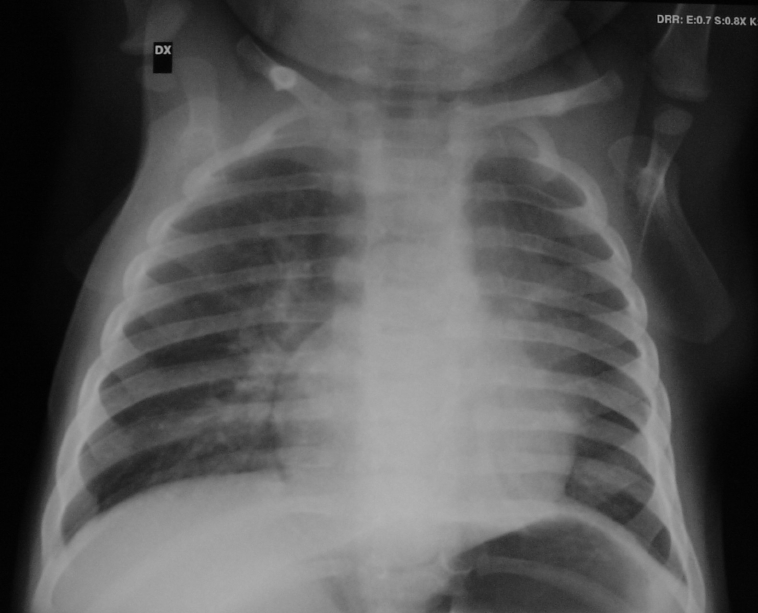
LRx
torace eseguito in corso di uno di questi episodi aveva evidenziato
la presenza di polmonite basale sinistra (Figura
1).
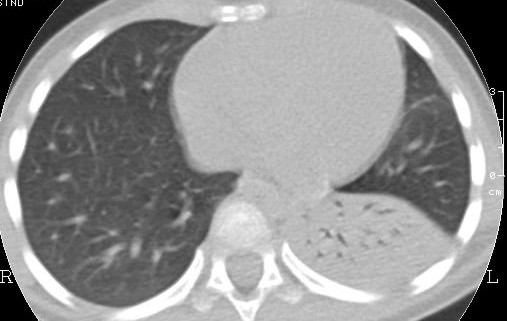
La TAC,
eseguita a risoluzione del terzo episodio infettivo, metteva in luce
la presenza di lesione malformativa, altrimenti non visibile in
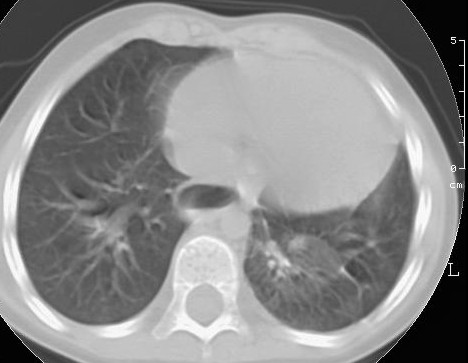
radiologia convenzionale (Figura 2).
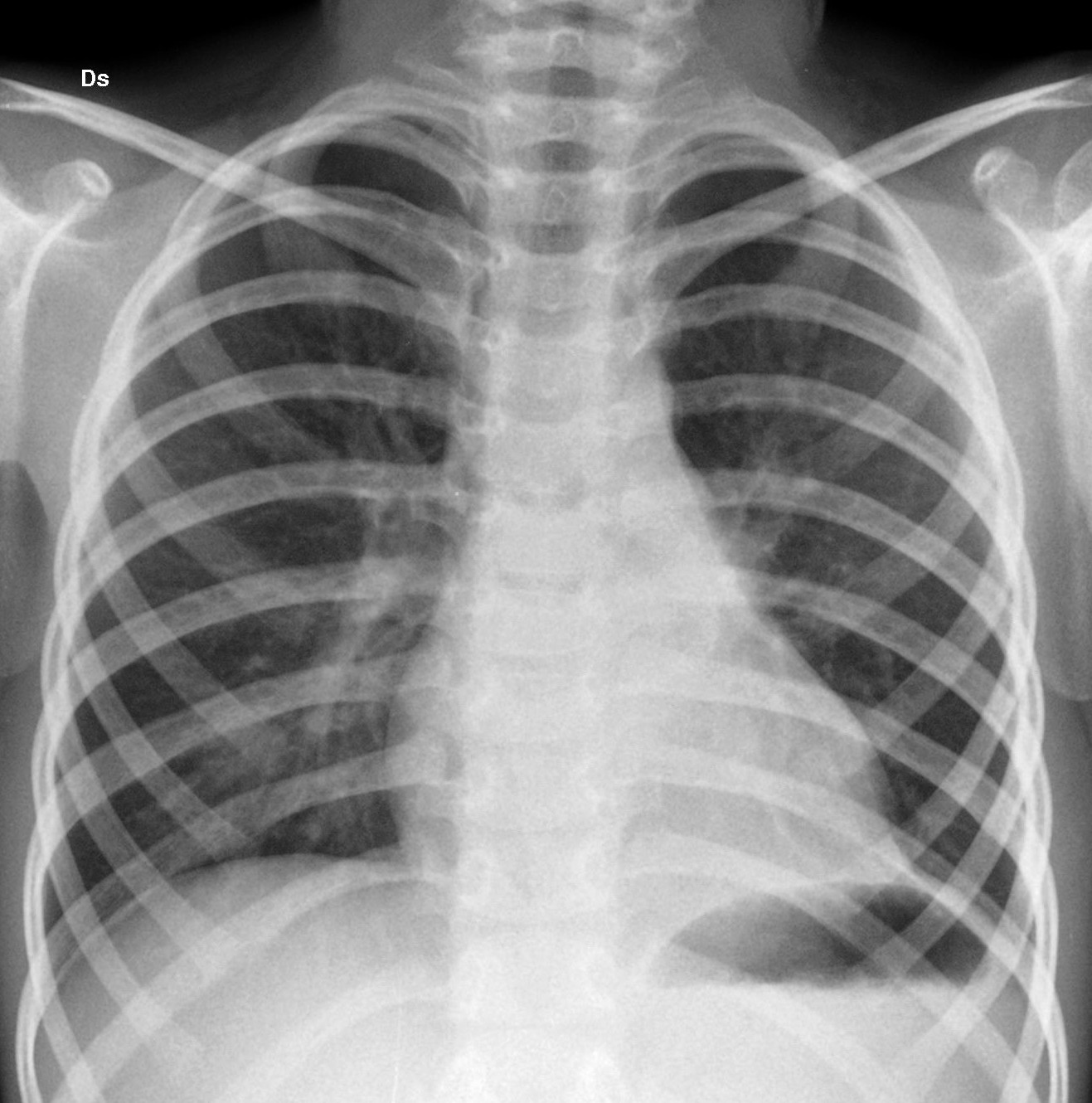
Alletà
di 4 anni il bambino veniva sottoposto a intervento di lobectomia
inferiore sinistra per via toracoscopica.
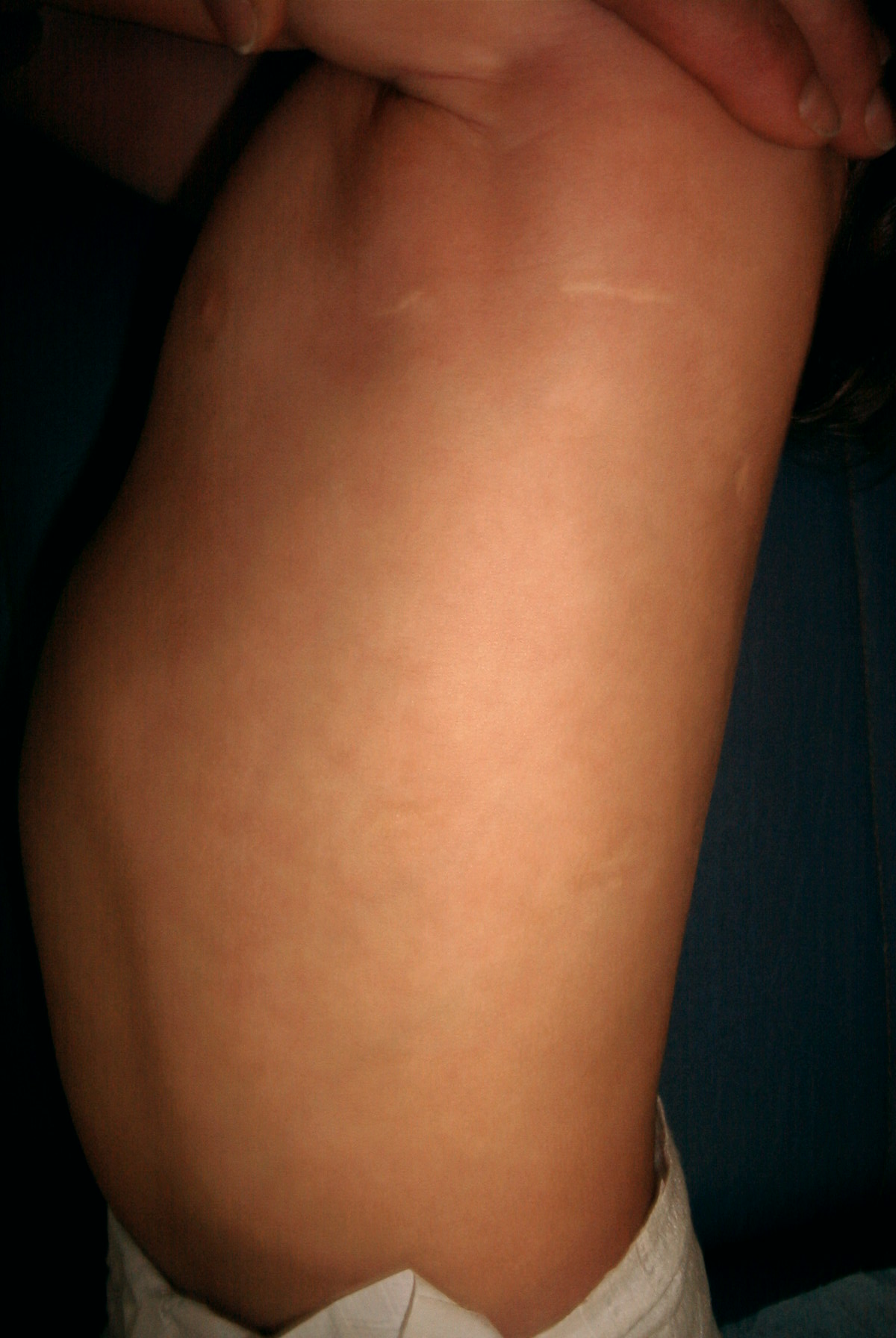
In Figura
3 limmagine del controllo a un anno dallintervento.
Figura
1
Figura
2
Figura
3
Caso
2
R. è
una bambina di 4 anni in cui la diagnosi di malformazione polmonare
era stata posta alletà di 3 anni, come reperto tardivo,
secondario a episodi broncopneumonici basali a sinistra. In tale
occasione era stata evidenziata per la prima volta lesione
microcistica basale sinistra associata ad area enfisematosa
perilesionale (Figura 4).
Veniva
eseguito lintervento chirurgico di segmentectomia basale
sinistra per via toracoscopia. A 6 mesi dallintervento era
ricomparsa polmonite basale sinistra sulla regione enfisematosa. Il
ripetersi di episodi infettivi aveva richiesto, a distanza di due
anni dal primo intervento, lasportazione dellintero
lobo inferiore sinistro.
La Figura
5 mostra controllo al un mese dopo il secondo intervento di
lobectomia eseguito per via toracoscopia.
Figura
4
Figura
5
Caso 3
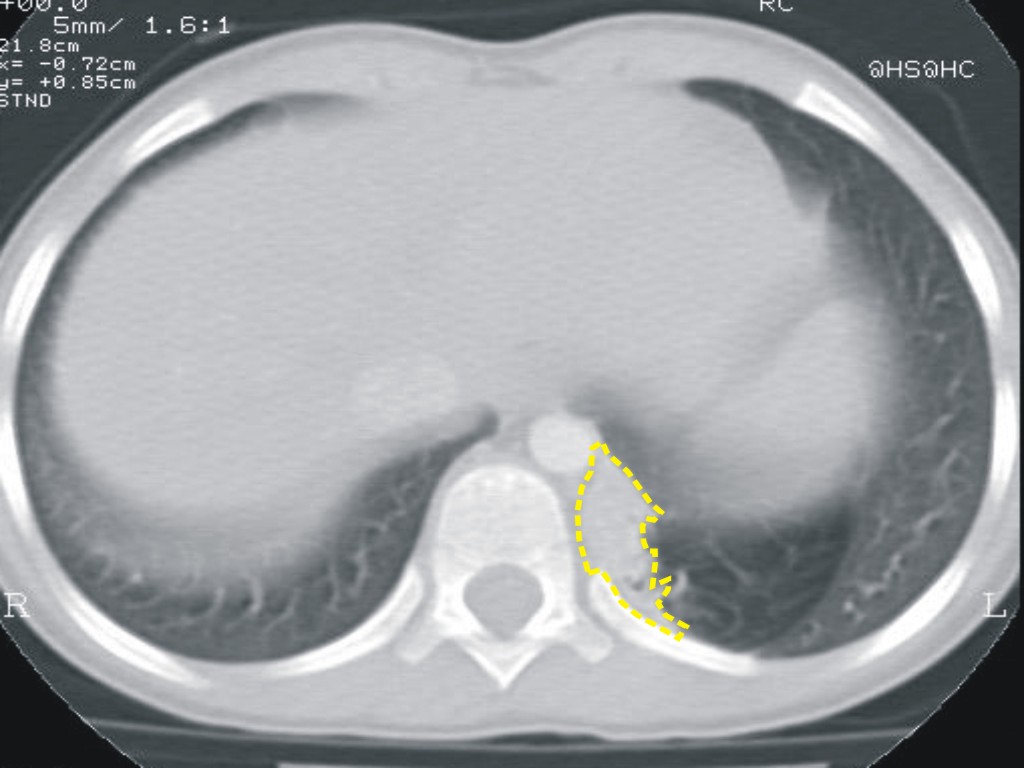
L. è
un neonato di un giorno di vita e presenta distress respiratorio
secondario a deviazione del mediastino da massa toracica sinistra. In
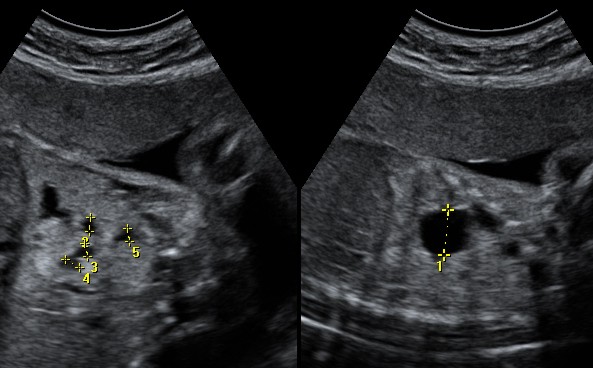
diagnosi prenatale veniva individuata una lesione macrocistica
compatibile con CAM tipo 1. Nel corso del secondo trimestre di
gravidanza si assisteva a comparsa di deviazione mediastinica, con
peggioramento della compressione a fine gravidanza (Figura
6)
In
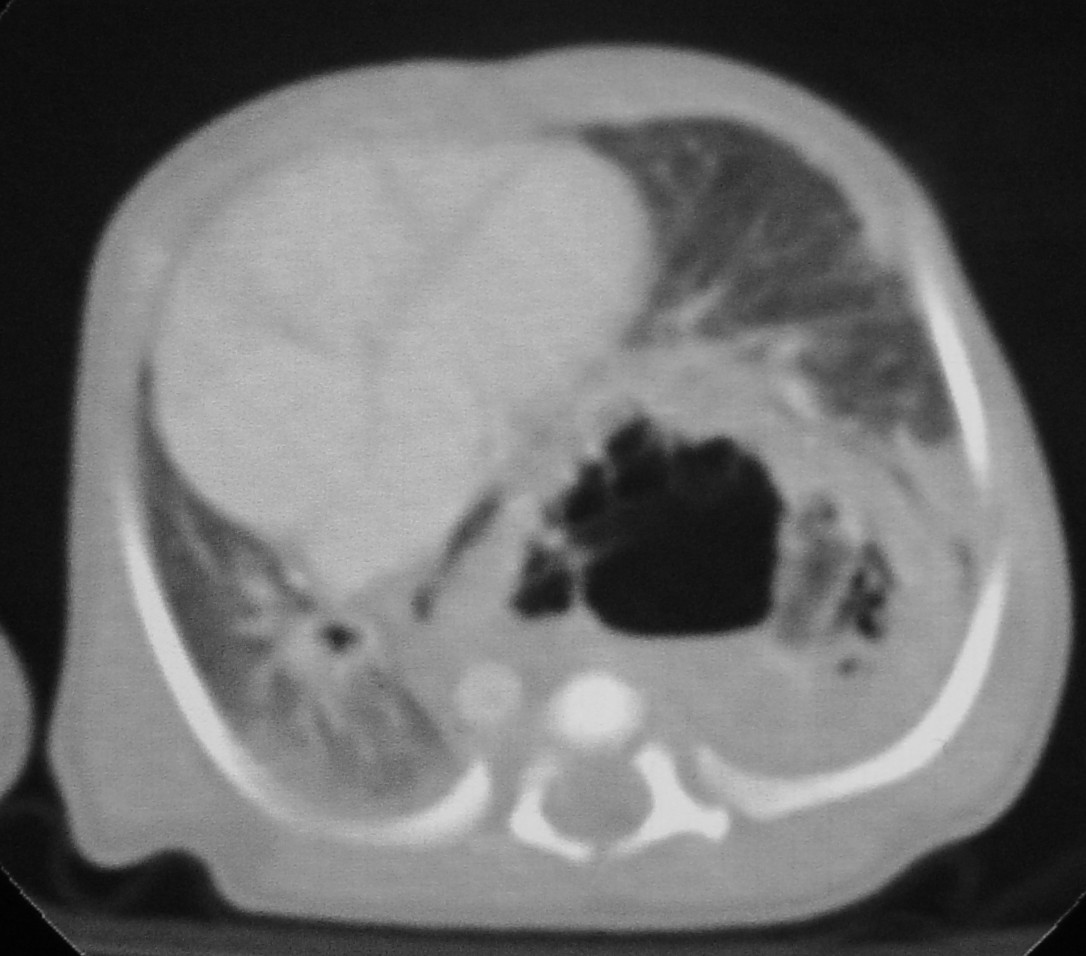
seconda giornata di vita veniva eseguita una lobectomia superiore
sinistra. Il decorso è stato privo di complicanze (Figura
7 e Figura 8). Controllo post operatorio a
1 mese (Figura 9).
Figura
6
Figura
7
Figura
8
Figura
9
La
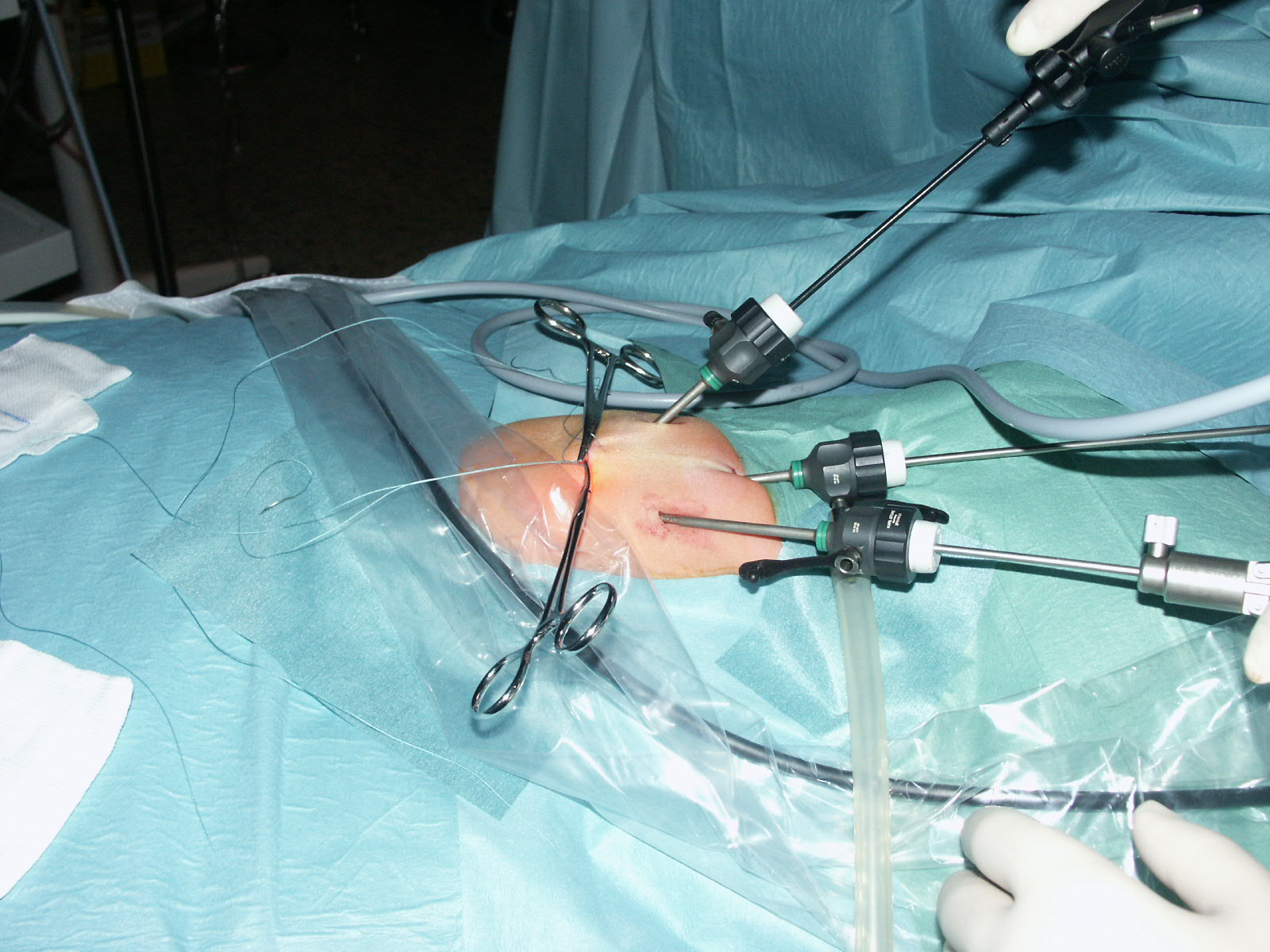
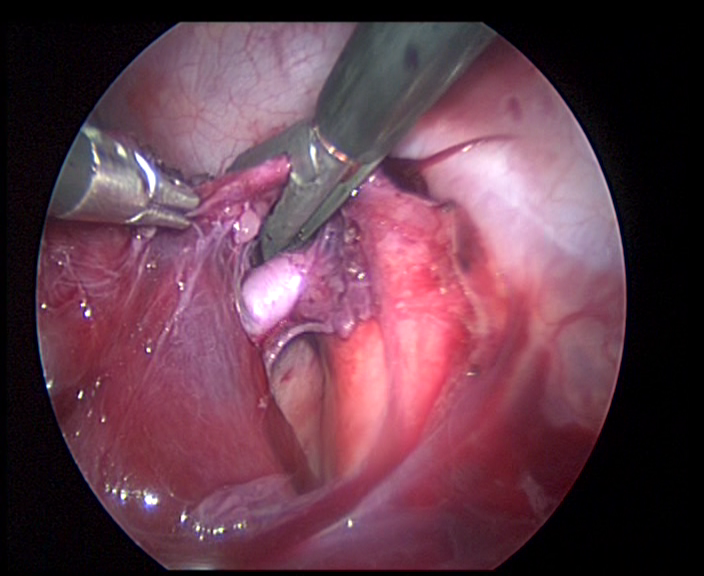
chirurgia mini invasiva
Figura
10. Toracoscopia13
Figura
11. Toracoscopia
Figura
12. Aspetto toracoscopio di resezione lobare inferiore destra.
Figura
13: Risultato cosmetico a 1 anno.
Da
ricordare
Leco
fetale definisce la storia della malformazione e la prognosi
neonatale. Per i casi complessi ha il compito di mettere a punto un
piano adeguato di assistenza perinatale14,15.
Lindagine
radiologica toracica standard non definisce il tipo di lesione,
spesso non identifica neppure la presenza della lesione. LRx
torace individua i segni indiretti della presenza della lesione e la
presenza di lesioni a grosse bolle.
In tutti
i pazienti la diagnosi definitiva postnatale necessita della
metodica TAC.
Nei
pazienti asintomatici e con diagnosi prenatale è
consigliabile eseguire la TAC nei primi mesi di vita, anche quando
la lesione è scomparsa durante gli ultimi controlli
ecografici prenatali.
La
terapia è chirurgica, possibilmente entro lanno di
vita, per evitare la comparsa di fenomeni infiammatori e favorire
unadeguata crescita del polmone residuo16,17,18.
Bibliografia
Cloutier
MM, Schaeffer DA, Hight D. Congenital
cystic adenomatoid malformation. Chest 1993;103:761-4.
Bratu
I, Flageole H, Chen MF, Di Lorenzo M, Yazbeck S, Laberge JM. The
Multiple Facets of Pulmonary malformations..J
Pediatr Surg2003;12:17-37.
Kunisaki
SM, Barnewolt CE, Estroff JA, et al. Large fetal congenital
adenomatoid malformations: growth trends and patient survival.
Journal of Paediatric Surgery 2007;42:404-10.
Langston
C. New Concepts in the Pathology of Congenital Lung Malformations.
Seminars in Paediatric Surgery 2003;12:17-37.
Adzick
NS, Flake AW, Crombleholme TM. Management of Congenital Lung
Lesions. Seminars in Paediatric Surgery 2003;12:10-6.
Dommergues
M, Louis-Sylvestre C, Mandelbrot L, et al.
Congenital adenomatoid malformation of the
lung: when is active fetal therapy indicated? Am
J Obstet Gynecol
1997;177:953-8.
Aziz D,
Langer JC, Tuuha SE, Ryan G, Ein SH, Kim PC. Perinatally diagnosed
asymptomatic congenital cystic adenomatoid malformation: to resect
or not? J Pediatr Surg 2004;39:329-34.
Miniati
DN, Chintagumpala M, Langston C, et al. Prenatal presentation and
outcome of children with pleuropulmonary blastoma. J Pediatr Surg
2006;41:66-71.
Tawill
MI, Pilling DW. Congenital cystic adenomatoid malformation: is there
a difference between the antenatally and postnatally diagnosed
cases? Pediatr Radiol 2005;35:39-40.
Fraggetta
F, Cacciaguerra, Nash R, Davenport M. Intra-abdominal Pulmonary
Sequestration Associated with Congenital Cystic Adenomatoid
Malformation of the Lung: Just an Unusual Combination of Rare
Pathologies? Pathol Res Pract 1998;194:209-11.
Conran
RM, Stocker JT. Extralobar sequestration with frequently associated
congenital cystic adenomatoid malformation, type II: report of 50
cases. Pediatr Dev Pathol 1999;2:454-63.
Cano I,
Anthon-Pacheco JL, Garcìa A, Rothenberg S. Video-assisted
thoracoscopic lobectomy in infants. European Journal of
Cardio-thoracic Surgery 2006;29:997-1000.
Sauvat
F, Michel JL, Benachi A, Edmond S, Revillon Y. Management of
asymptomatic neonatal cystic adenomatoid malformations. J Pediatr
Surg 2003;38:548-52.
Tsao K,
Albanese CT, Harrison MR. Prenatal Therapy and Mediastinal Lesions.
World J Surg 2003;27:77-83.
Van
Leeuwen K, Teitelbaum DH, Hirschl RB, et al. Prenatal Diagnosis of
Congenital Cystic Adenomatoid Malformation and Its Postnatal
Presentation. Surgical Indications, and Natural History. J Pediatr
Surg 1999;34:794-9.
Usui N,
Kamata S, Sawai T, et al. Outcome Predictors for Infants WITH Cystic
Lung Disease. J Pediatr Surg 2004;39:603-6.
Laberge
JM, Flageole H, Pugash D, et al. Outcome of prenatally diagnosed
congenital cystic adenomatoid lung malformation: A Canadian
experience. Fetal Diagn Ther 2001;16:178-86.