Marzo 2009 - Volume XII - numero 3
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Alopecia
bitemporale e strie di iperpigmentazione in lattante con ritardo
psicomotorio
1SC
Neonatologia e Terapia Intensiva Neonatale, 2SC Genetica
Medica IRCCS "Burlo Garofolo", Trieste
3attualmente
SC Centro Coordinamento Regionale Malattie Rare, Azienda
Ospedaliero-Universitaria Santa Maria della Misericordia, Udine
Indirizzo
per corrispondenza: ciana.giovanni@aoud.sanita.fvg.it
BITEMPORAL
ALOPECIA AND STREAKS OF HYPER-PIGMENTATION IN A INFANT WITH
MENTAL RETARDATION
Key
words
Pallister-Killian
syndrome, 12p tetrasomy, Iroquois Indians hair, Hyper
pigmentation lines
Summary
The
careful classification of a psychomotor delay in an infant is
often a difficult task for paediatricians. Many hospital
admissions for clinical, instrumental and laboratori
investigations, are often needed, even though an exact diagnosis
is not always reached. In some cases, however, the presence of a
few very distinctive clinical features may allow for a prompt
diagnosis and induce the paediatrician to choose the best
diagnostic tools to confirm the clinical picture. We report one
case of Pallister-Killian syndrome with mental retardation,
streaks of hyper-pigmentation, and facial anomalies, including
prominent forehead with sparse anterior scalp hair. |
CASO
CLINICO
Il
piccolo nasce a 37 settimane di età gestazionale da TC di
emergenza per movimenti fetali ridotti. In gravidanza, segnalato
polidramnios non spiegato dalla 35° settimana. Alla nascita,
ricoverato in Neonatologia per presenza di distress respiratorio, poi
rapidamente risoltosi, Apgar 9 e 10, parametri antropometrici nella
norma.
Durante
il ricovero vengono riscontrati i seguenti problemi: un
ipoparatiroidismo transitorio risoltosi lentamente con dosaggi
gradualmente aumentati di calcio gluconato e Vitamina D, un ipertono
assiale con arti superiori in flessione, tendenza a mantenere le mani
chiuse e difficoltà nell'evocazione dei riflessi neonatali.
Evidenti ancora un soffio sistolico cardiaco con riscontro
all'ecocardiografia di insufficienza della valvola mitrale e una
ipovalidità della suzione con necessità di integrazione
con gavage fino alla 17° giornata di vita.
Vista la
compresenza di ipoparatiroidismo, atteggiamento posturale non
convincente e cardiopatia congenita si è ipotizzata la
sindrome delezione 22q11.2. L'analisi FISH (fluorescence in situ
hybridization) eseguita su sangue periferico non ha messo in
evidenza tale delezione; inoltre l'analisi del cariotipo eseguita
sullo stesso tessuto è risultata normale. Rispetto alla
ipocalcemia, viene esclusa una sindrome Di George (presenza del timo
all'Rx torace). L'aspetto neurologico (atteggiamento posturale,
suzione ipovalida, ridotti movimenti fetali) è stato indagato
tramite RMN encefalo risultata nella norma per età. Dimesso,
durante il follow up si rende progressivamente evidente una ipotonia
con ritardo psicomotorio. All'esame obiettivo sono presenti
dimorfismi rappresentati da ipertelorismo con strabismo alternante,
fronte prominente , radice del naso piatta con narici anteverse,
filtro lungo con labbro superiore fine e labbro inferiore pieno,
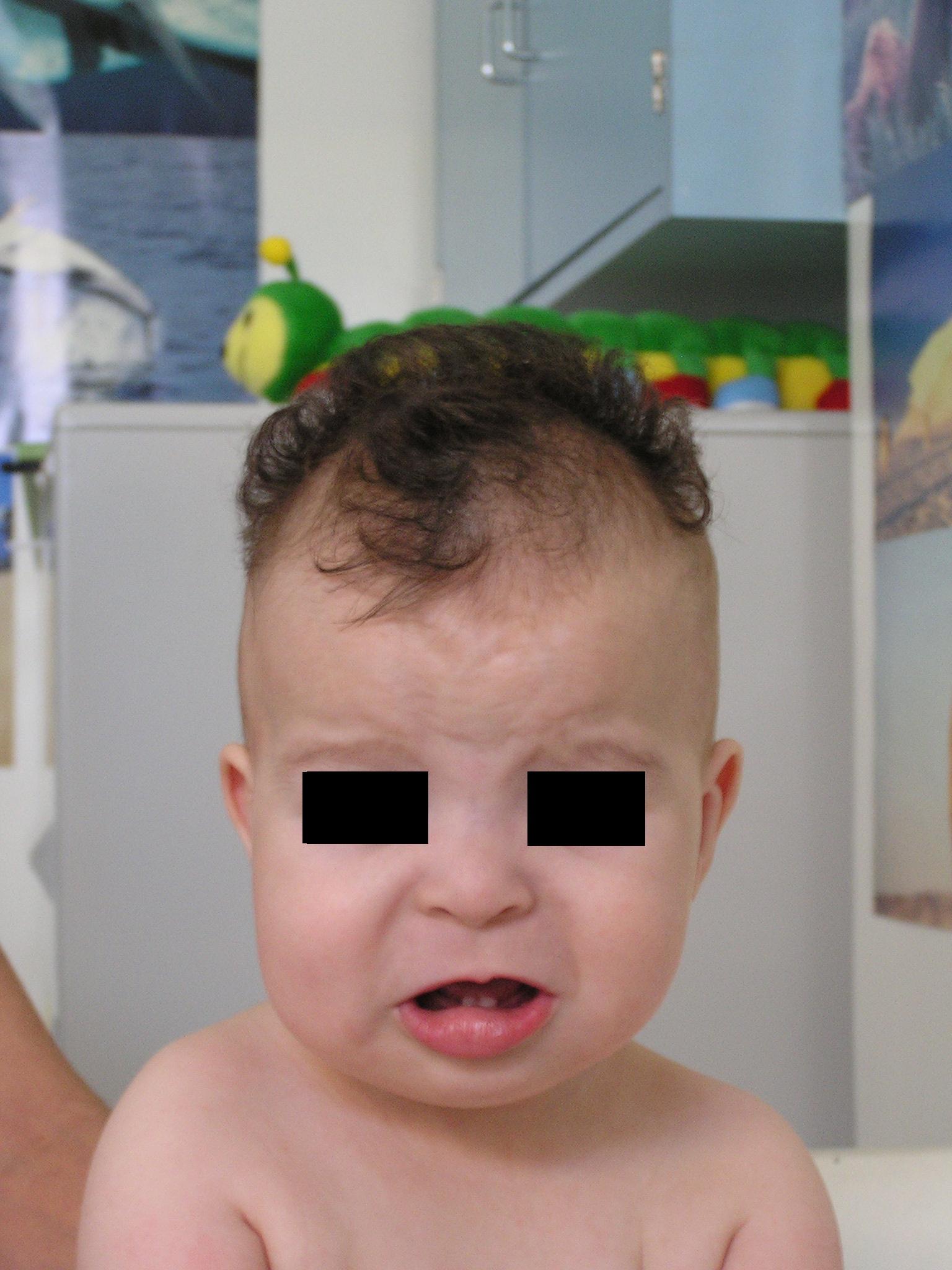
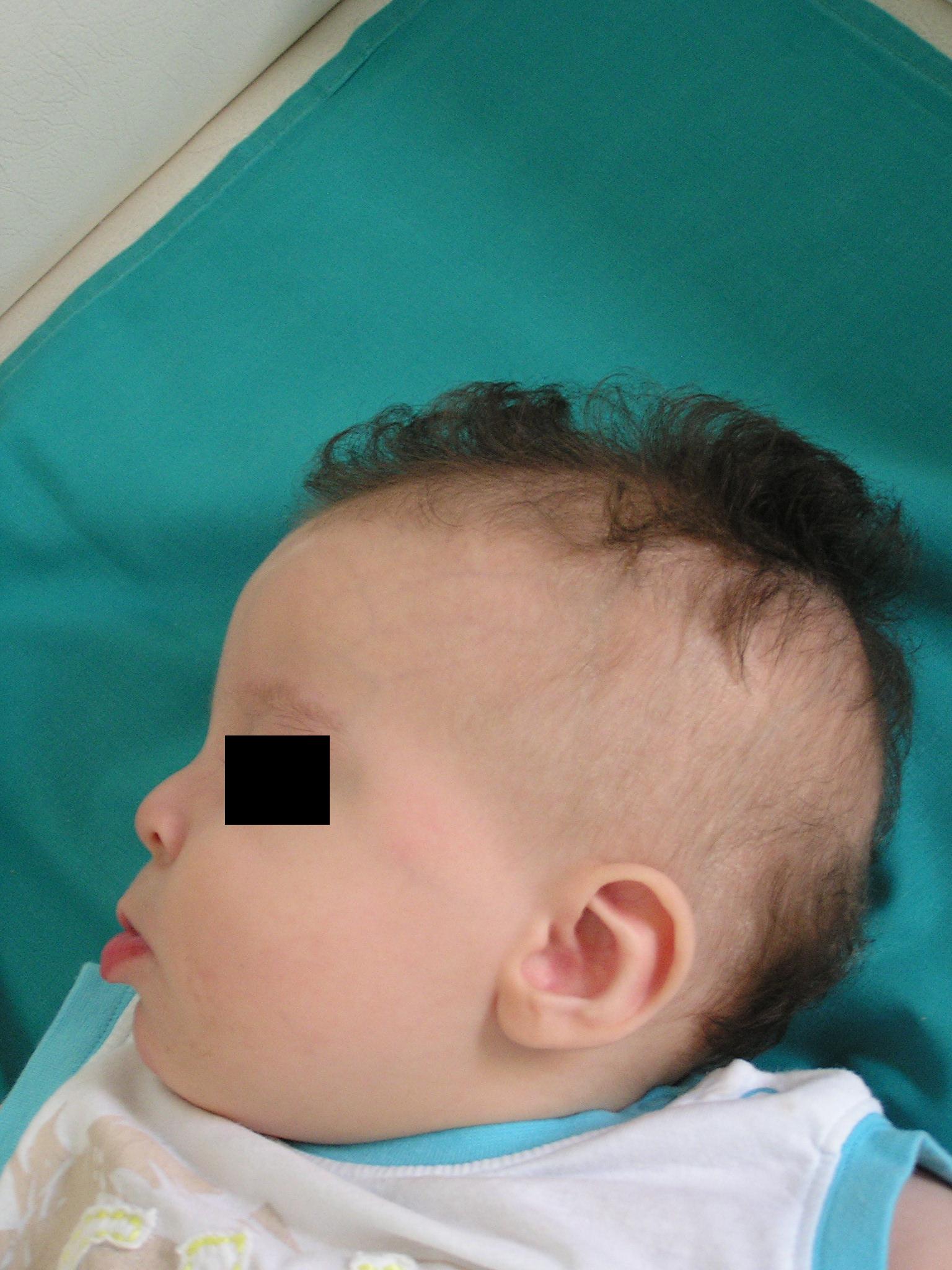
palato ogivale. Della facies colpisce soprattutto una particolare
rarefazione dei capelli sulle tempie (Figura 1
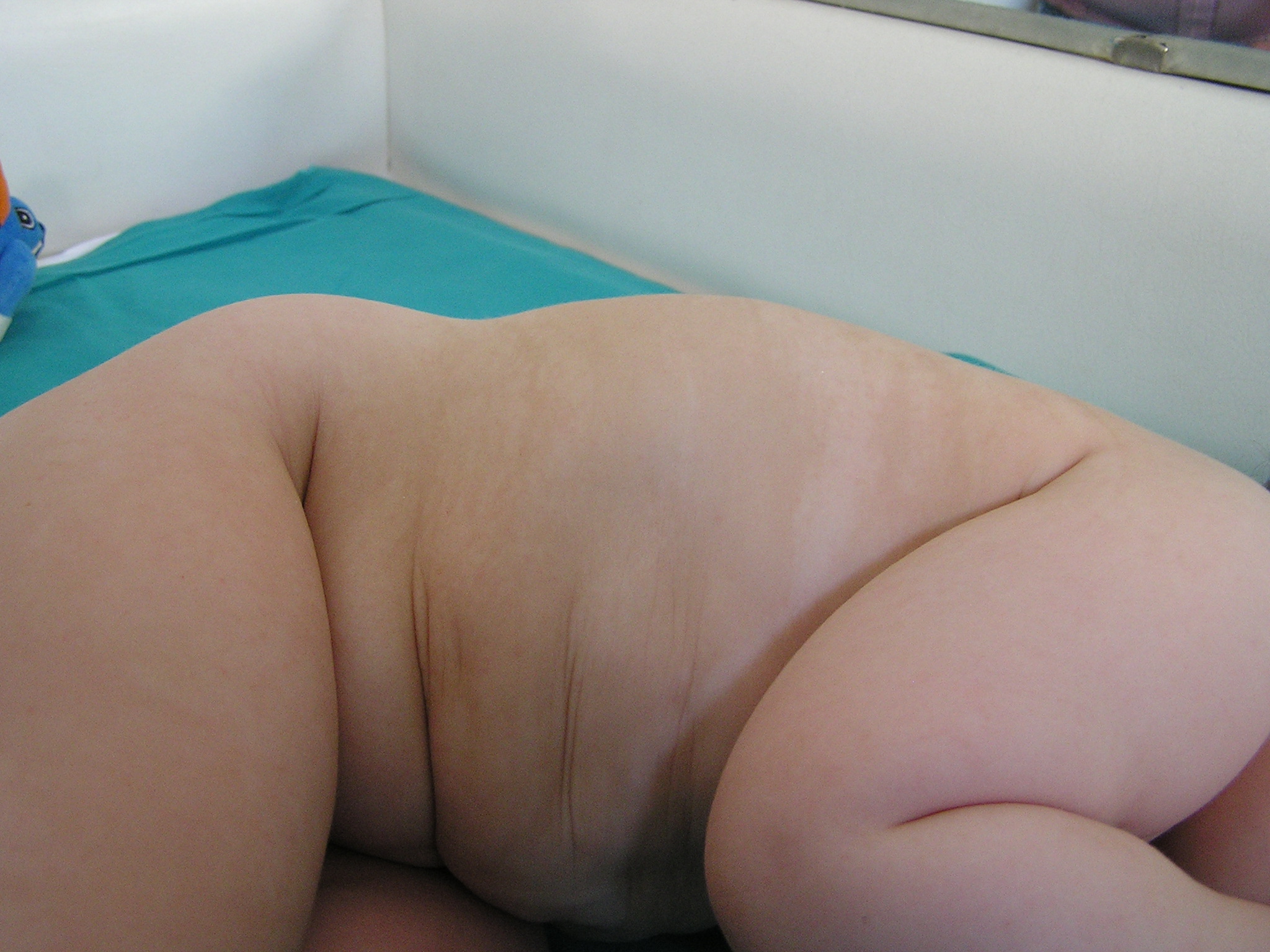
e Figura 2) mentre al tronco sono evidenti
delle strie di iperpigmentazione (Figura 3).
Questa
particolare alopecia bitemporale in primis, associata al ritardo
motorio ed alle anomalie della pigmentazione cutanea ci hanno fatto
sospettare la sindrome di Pallister-Killian.
E'
stata quindi effettuata una biopsia cutanea per coltura fibroblasti
sui quali è stata poi eseguita un'analisi del cariotipo che
ha messo in evidenza la presenza di un cromosoma soprannumerario in
seguito identificato come un isocromosoma del braccio corto del
cromosoma 12 [47,XY,i(12p)] nel 100% delle metafasi analizzate
confermando così tale sindrome.
Mediante
studio con micro satelliti per valutare l'origine parentale del
cromosoma sovra numerario, si è dimostrato che questo era di
origine materna .
Ulteriori
approfondimenti evidenziavano poi l'assenza di fenomeni critici
all'EEG, mentre un bilancio intellettivo veniva rinviato dopo
l'acquisizione degli occhiali per la correzione dello strabismo per
una più corretta esecuzione del test
DISCUSSIONE
La
sindrome di Pallister-Killian, o tetrasomia 12p a mosaico (presenza
di quattro copie del braccio corto del cromosoma 12), descritta per
la prima volta nel 19771, associa dismorfismi facciali
caratteristici, anomalie della pigmentazione, malformazioni
viscerali, ritardo mentale grave ed epilessia. A questi segni si
associa la presenza di un isocromosoma sovra numerario del braccio
corto del cromosoma 12, confinato nei fibroblasti cutanei.
L'isocromosoma è il risultato di un'aberrazione cromosomica
strutturale intracromosomica causata da un errore di divisione.
Il
cromosoma si rompe trasversalmente, anziché longitudinalmente,
a livello del centromero e, braccio lungo e braccio corto si
separano. Ognuno di essi costituirà un nuovo cromosoma
(l'isocromosoma appunto), in seguito uno dei due bracci viene perso,
in questo caso quello lungo.
Nella
sindrome di Pallister-Killian l'anomalia cromosomica è
relegata in alcuni tessuti, quali la cute, mentre i linfociti
presentano un cariotipo normale. Per tale motivo il sospetto
diagnostico trova conferma nell'analisi cromosomica effettuata su
fibroblasti ottenuti da biopsia cutanea. In epoca prenatale questa
anomalia cromosomica può essere evidenziata su amniociti ma
non su linfociti fetali.
Clinicamente,
il reperto più tipico alla nascita e nella prima infanzia è
l'alopecia bitemporale che associata all'abbondanza di capelli
presenti al vertice ricorda, come cita l'OMIM,
la capigliatura degli Indiani Irochesi.
A livello
prenatale i reperti ecografici più frequenti sono l'ernia
diaframmatica, accorciamento rizomelico degli arti ed il
polidramnios, anche se sono stati riportati difetti cardiaci,
polmonari, urogenitali, contratture articolari, idrope e
ventricolomegalia cerebrale. Recentemente è stato
sottolineato, come ulteriore reperto associato, un'aumentata
traslucenza nucale2 che peraltro è di per sè
un marker di molte anomalie cromosomiche di numero e struttura.
Nel
nostro caso la traslucenza nucale rientrava nei parametri normali ed
il test combinato del primo trimestre era risultato negativo. Per
tali motivi e per l'età materna non avanzata non era stata
proposta la diagnosi prenatale che avrebbe permesso di fare diagnosi
poiché l'isocromosoma del braccio corto del cromosoma 12 si
evidenzia negli amniociti.
Per
quanto riguarda gli altri possibili segni, alle 35 settimane di età
gestazionale l'ECOgrafia evidenziava un polidramnios tardivo senza
presenza però di note dismorfiche3.
Tra gli
altri reperti clinici evidenti nel nostro caso, erano presenti al
follow up delle strie cutanee di iperpigmentazione. Spot o strie di
iper- o ipopigmentazione sono presenti nel 43% dei casi, possono
rappresentare un marker clinico diagnostico importante nei casi di
fenotipo lieve4 e si rendono evidenti nel primo anno di
vita. Prima della descrizione completa di sindrome, alcuni di questi
casi venivano definiti come ipomelanosi di Ito con anomalie
addizionali.
Il
piccolo, fino all'età attuale di 14 mesi, non ha presentato
convulsioni; tali sintomi si riscontrano in circa il 40% dei casi,
possono comunque comparire anche tardivamente e in assenza di quadri
malformativi cerebrali.
Il
ritardo mentale in questa sindrome è di norma severo anche se
vengono riportate singole segnalazioni di casi con ritardo mentale
lieve o assente, nei quali la presenza dell'isocromosoma è
stata evidenziata in circa il 30% delle metafasi dei fibroblasti
cutanei4. Va peraltro sottolineato che non c'è
una netta correlazione tra percentuale di cellule con tetrasomia 12
nei fibroblasti cutanei e severità del quadro clinico. Nel
nostro paziente, anche se non precisamente quantificato, il ritardo
mentale sembra severo.
Nonostante
la tetrasomia 12p sia la più frequente tra le tetrasomie
autosomiche, l'incidenza della sindrome è bassa (inferiore a
1/10.000 nati) e la frequenza aumenta con l'avanzare dell'età
materna. L'origine della tetrasomia 12p è de novo e la
consulenza genetica, in caso di successiva gravidanza, può
essere molto rassicurante. Come messaggio pratico importante, il
nostro caso insegna che una anomalia cromosomica può essere
presente in cellule diverse dai linfociti periferici e che il compito
del clinico esperto è di interfacciarsi e di guidare il
laboratorio di citogenetica nella scelta del tessuto giusto da
indagare.
Bibliografia
1.
Pallister PD, Mesiner LF, Elejade BR et al. The Pallister mosaic
syndrome. Birth defects 1977; XIII (3B):103-10
2.
Escribano Abad D, Arbuès Gabarre J, Izquierdo AM.
Pallister-Killian Syndrome presenting with a complex congenital heart
defect and increased nuchal translucency. Ultrasound Med.
2006;25:1475-1480.
3.
Paladini D, Borghese A, Arienzo M, Teodoro A, Martinelli P, Nappi C.
Prospective ultrasound diagnosis of Pallister-Killian syndrome in the
second trimester of pregnancy: the importance of the fetal facial
profile. Prenat Diagn 2000;20:996-998.
4.
Genevieve D, Cormier-Daire V, Sanlaville D, et al. Mild phenotype in
a 15-year-old boy with Pallister-Killian syndrome. Am J Med Genetics
2003; 116A:90-93
Vuoi citare questo contributo?