Maggio 2008 - Volume XI - numero 5
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Lesione
nodulare retroauricolare in un lattante. Descrizione di un caso
clinico e revisione della letteratura
1Clinica
Pediatrica, Università degli studi di Ferrara; 2Istituto
di Radiologia, Università degli studi di Ferrara; 3Istituto
di Anatomia e Istologia Patologica, Università degli studi di
Padova
Indirizzo
per corrispondenza: elisagiaco@gmail.com
Retroauricolar
nodular mass in a 5 month-old infant. A case report
Key
words
Infant,
skull lesions, myofibroma, differential diagnosis
Summary
We
reported a 5 month-old infant case, admitted to our institution
for a retroauricular mass. We described our diagnostic approach
and the differential diagnosis that was progressively excluded.
Lesions of the skull were present as lumps on the head, and by a
broad differential diagnosis they could be referred to as
inflammatory, malformative, traumatic and neoplastic lesions, so
clinical presentations and diagnostic imaging could be useful. In
most of cases, biopsy is necessary to make a definitive
diagnosis. |
M.E.
giunge alla nostra osservazione all'età di 5 mesi, per la
recente comparsa di una tumefazione in sede retroauricolare destra.
All'esame obiettivo la tumefazione si presenta di consistenza
solida, di circa 2 cm di diametro, non dolente alla palpazione e con
cute sovrastante integra. La restante obiettività è
negativa. L'ecografia mostra una massa disomogeneamente
iperecogena, non nettamente delimitata dall'osso temporale
sottostante, con modica e omogenea vascolarizzazione al
color-doppler.
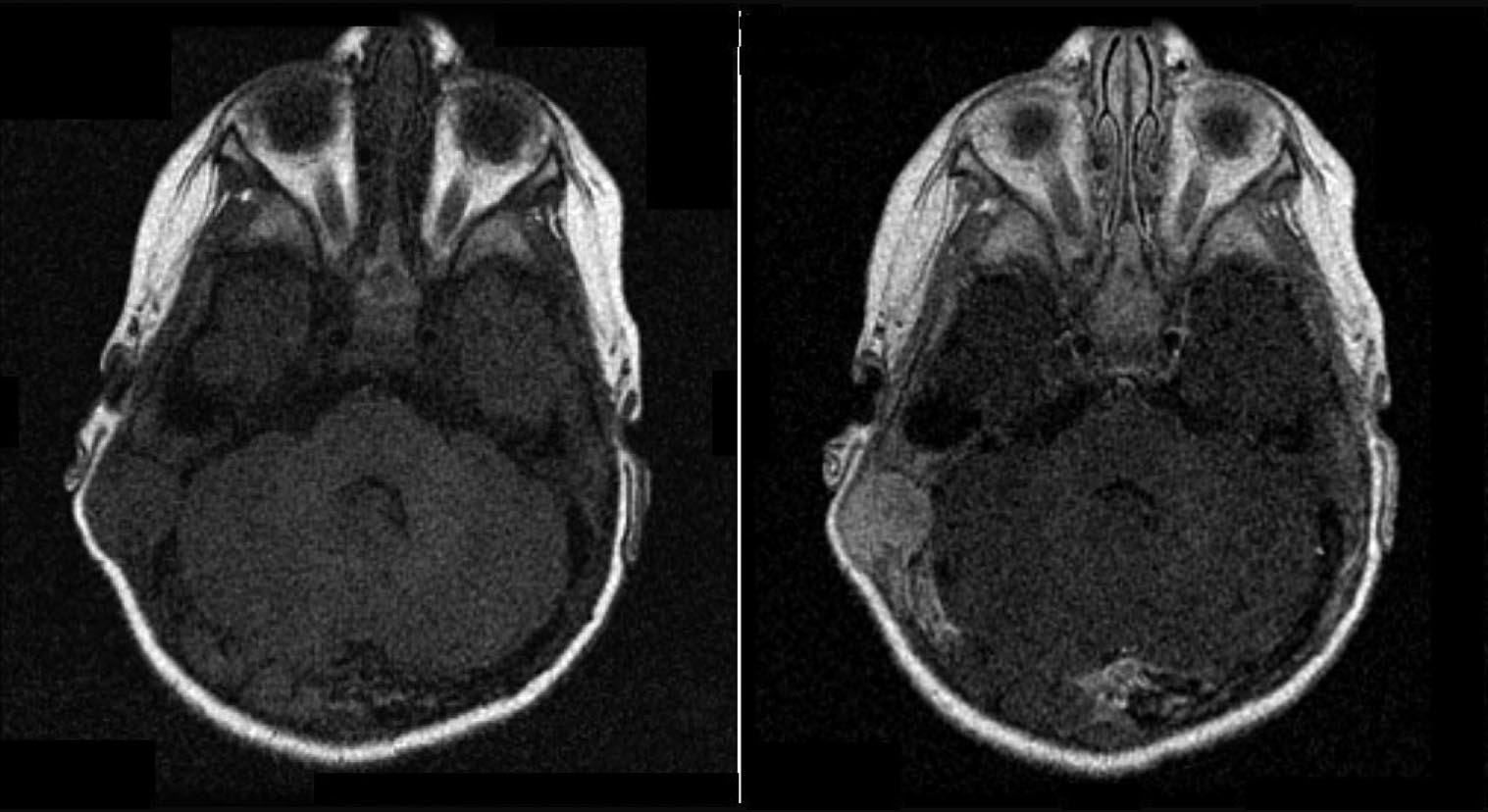
A questo
punto viene eseguita una Risonanza Magnetica Nucleare (RMN) per
definire l'eventuale coinvolgimento dell'encefalo: si documenta
una lesione che determina un'ampia osteolisi a tutto spessore della
teca cranica, giungendo a contatto con il cervelletto (Figura
1 e Figura 2). La Tomografia
Computerizzata (TC) conferma il reperto (Figura
3).
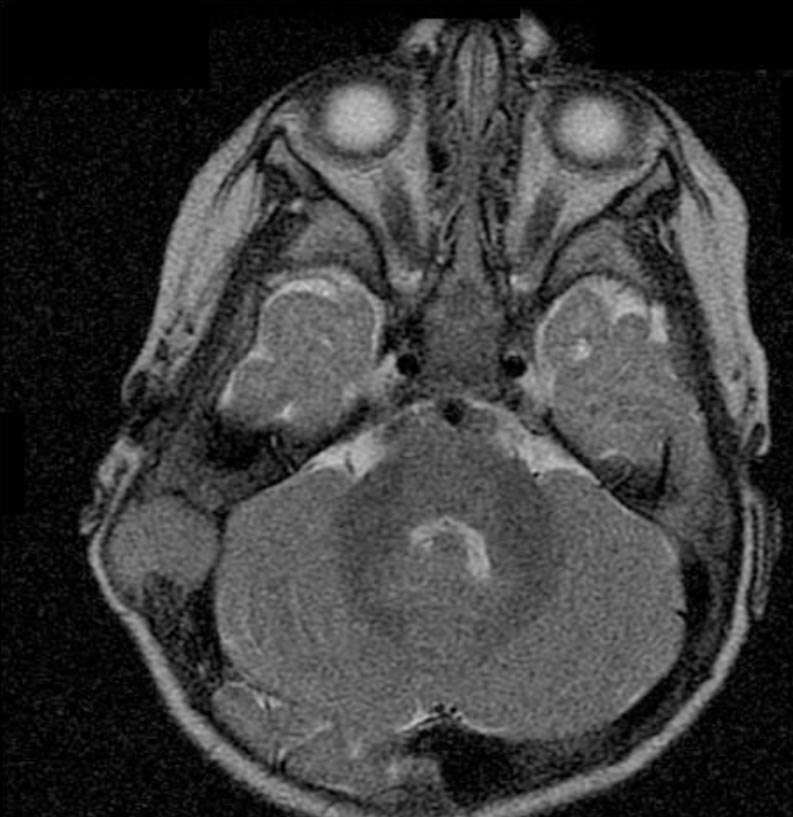
Figura
2. Immagine alla RMN assiale dipendente dal T2. Il tessuto
patologico, isointenso, rispetto alla sostanza grigia prende rapporti
con la superficie cerebellare.
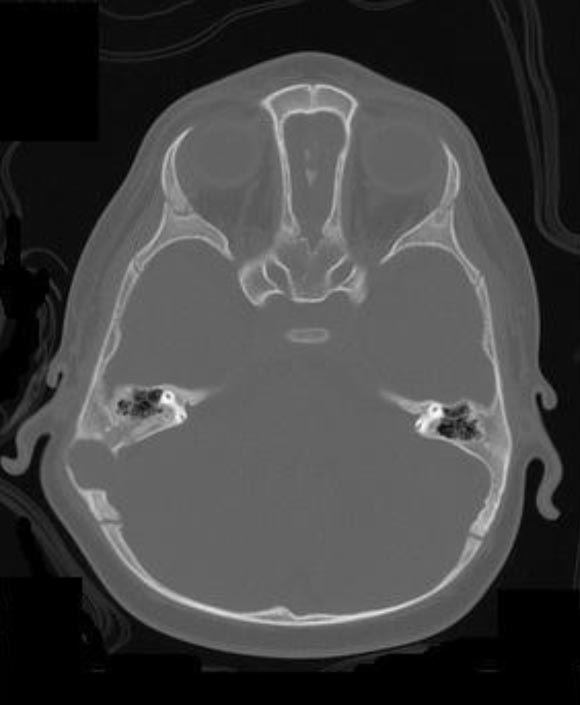
Le
tumefazioni a carico del capo possono essere di origine
infiammatoria, malformativa, traumatica e neoplastica. La sede, la
consistenza teso-elastica, la mobilità sui piani superficiali
e profondi e il dolore alla palpazione suggeriscono una
linfoadenopatia infiammatoria, che delle tumefazioni rappresenta la
causa più frequente. Nel nostro paziente invece la tumefazione
era di consistenza dura e apparentemente non dolente, con mobilità
scarsa rispetto ai piani superficiali e profondi, per cui questa
ipotesi è apparsa subito poco probabile.
Ecografia
e Rx sono le indagini di primo livello nella diagnosi differenziale
della natura della lesione, che spesso necessita di approfondimento
mediante TC e RMN per valutare i rapporti con le ossa craniche e
l'encefalo.
Nel
nostro caso l'esame ecografico ha documentato una massa
iperecogena, disomogenea, con interessamento dell'osso temporale;
l'Rx non è stata eseguita; TC e RMN hanno mostrato una
protrusione della massa verso l'encefalo.
Le
lesioni delle ossa craniche possono rappresentare la localizzazione
secondaria di una patologia neoplastica (metastasi). Tra le neoplasie
maligne della prima infanzia le più importanti da considerare
sono il neuroblastoma e i linfomi.
Abbiamo
eseguito la ricerca dei metaboliti delle catecolamine urinarie e
dell'enolasi neurono-specifica plasmatica, che è risultata
negativa; questo non permette di escludere con certezza che si tratti
di un neuroblastoma metastatico, ma lo rende meno probabile.
Altre
diagnosi differenziali che devono essere considerate sono:
- Linfomi: generalmente non danno lesioni litiche a livello osseo anche se sono stati descritti alcuni casi in letteratura1.
- Istiocitosi a cellule di Langerhans: è una malattia proliferativa che può presentarsi con lesioni intraossee litiche solitarie o multiple, a livello del cranio o di qualunque altro segmento osseo. La lesione solitaria (granuloma eosinofilo) coinvolge più spesso il cranio2, si riscontra con maggiore frequenza in bambini di età compresa tra i 5 e i 10 anni e può costituire un reperto radiografico accidentale. Le lesioni multiple sono più frequenti nei bambini più piccoli e radiologicamente conferiscono al cranio un aspetto a carta geografica. Al microscopio le lesioni sono costituite da cellule di Langerhans con abbondante citoplasma eosinofilo e infiltrato infiammatorio di tipo cronico (linfociti e plasmacellule). Il granuloma eosinofilo alla RMN appare frequentemente isointenso rispetto al tessuto cerebrale sia nelle immagini T1 dipendenti che in quelle T2 dipendenti come nel caso del nostro paziente. Anche l'età del paziente e l'obiettività clinica sono compatibili con questa patologia.
- Cisti epidermoidi e cisti dermoidi: sono lesioni benigne, di derivazione epidermica, nella maggior parte dei casi congenite. Possono localizzarsi in sede sottocutanea e intraossea, e in tal caso le ossa coinvolte sono più frequentemente le ossa craniche, in particolare l'osso frontale, il temporale e il parietale. In una recente revisione le cisti epidermoidi sono risultate le più frequenti lesioni primitive delle ossa craniche, seguite dal granuloma eosinofilo3. Radiologicamente mostrano margini netti e ben definiti, ma a volte provocano erosione litica dell'osso interessato per crescita espansiva. Possono estendersi in profondità e comprimere l'encefalo. Al microscopio sono costituite da cellule squamose cheratinizzate; le cisti dermoidi presentano anche elementi annessiali (follicoli piliferi, ghiandole sebacee). Il trattamento consiste nell'escissione della lesione, che generalmente non recidiva. Questa diagnosi è compatibile con il nostro caso.
- Meningioma: è un tumore che si sviluppa dall'aracnoide e ha crescita lenta. Nonostante sia molto raro in questa fascia di età, il suo aspetto alla RMN è sovrapponibile a quello del piccolo paziente4.
- Miofibroma: è un tumore mesenchimale benigno, solitario o multicentrico. I tumori si riscontrano più frequentemente nel sottocute, ma possono coinvolgere ogni organo, e sono state descritte anche lesioni ossee primitive3. Oltre il 50% dei pazienti con lesioni solitarie e oltre il 90% di quelli con lesioni multiple presentano le manifestazioni della malattia già alla nascita o nei primi mesi di vita. I miofibromi si trovano più frequentemente a livello della testa e del collo o nel tronco5. Il fibrosarcoma è l'equivalente maligno, tende ad invadere i tessuti circostanti e dà metastasi a distanza. Il fibrosarcoma congenito è il più comune sarcoma sotto l'anno di vita, presenta un basso grado di malignità e metastatizza più raramente4. Per l'obiettività clinica e per le caratteristiche all'imaging, anche queste diagnosi sono compatibili con il nostro caso.
- Osteoblastoma: è un raro tumore osseo, che si presenta più frequentemente nelle vertebre e nelle ossa lunghe e raramente nel cranio6,7. La fascia di età più colpita è quella tra i 10 e i 25 anni. Nonostante sia un tumore benigno, è localmente distruttivo. Le caratteristiche radiologiche sono aspecifiche. Questa diagnosi appare poco probabile.
- Neurofibroma è in genere parte di un quadro clinico ben definito e nelle immagini RMN di solito, si presenta iperintenso nelle sequenze T2 dipendenti. Questa diagnosi appare alquanto improbabile, dato che il bambino è molto piccolo e non ha macchie caffè-latte, che sono generalmente il primo segno di NF1.
- Emangioma cavernoso intraosseo: gli emangiomi rappresentano lo 0,7% dei tumori ossei primitivi3; tuttavia la loro presentazione caratteristica è quella cranica, in particolare a livello dell'osso frontale, e quella vertebrale, probabilmente per la ricca vascolarizzazione venosa di queste sedi8. Da una recente classificazione basata sul comportamento clinico e sulle caratteristiche endoteliali, gli emangiomi devono essere distinti dalle malformazioni vascolari; l'emangioma cavernoso deve essere riclassificato come malformazione venosa9,10. Esso può crescere erodendo l'osso e comprimendo il tessuto nervoso circostante. L'ecocolor-doppler permette di escludere questa diagnosi.
Poiché
le indagini radiologiche non permettono di porre diagnosi, viene
eseguita exeresi della neoformazione e l'esame istologico mette in
evidenza un quadro caratterizzato dalla contemporanea presenza di
miofibroblasti (come nel miofibroma) e di cellule fusate disposte a
spina di pesce (come nel fibrosarcoma), secondo un pattern che viene
definito bifasico. L'esame immunoistochimico evidenzia la
positività all'actina muscolare, caratteristica del
miofibroma, solo nelle aree miofibroblastiche.
Dato che
dal solo esame istologico non è possibile escludere la
malignità della lesione, viene eseguita sul pezzo bioptico
l'indagine molecolare RT-PCR, atta a identificare il trascritto
ETV6-NTRK3, dovuto alla traslocazione t(12;15)(p13;q25), che è
specifica del fibrosarcoma congenito. L'indagine risulta negativa,
permettendoci di formulare diagnosi di miofibroma.
Eziologia
e patogenesi
La
Miofibromatosi Infantile (MI) è un raro disordine
dell'infanzia, a eziopatogenesi sconosciuta, descritta per la prima
volta nel 195111. Si caratterizza per la presenza di singoli o
multipli miofibromi (tumori mesenchimali benigni) di dimensioni
comprese tra 1 e 7 cm. è più frequente nei maschi
(M:F=2,4:1)5-12. La presentazione è generalmente
sporadica, anche se sono descritti casi familiari a probabile
ereditarietà autosomica dominante13, con decorso
più severo e per i quali è raccomandata la consulenza
genetica14,15.
Clinica
Le
manifestazioni cliniche e la prognosi dipendono dal numero e dalla
sede delle lesioni. Si distinguono 3 forme di MI:
- MI solitaria
- MI multicentrica senza coinvolgimento viscerale
- MI generalizzata con coinvolgimento viscerale5,16-18
La storia
naturale delle prime due forme è caratterizzata da un primo
periodo di rapida crescita nella forma solitaria e di aumento del
numero delle lesioni nella forma multicentrica, seguito da una fase
di stabilizzazione e successivamente da una regressione spontanea
che, soprattutto nella forma solitaria, avviene entro 1-2 anni dalla
diagnosi19 e nell'80% dei casi non recidiva5.
La forma con coinvolgimento viscerale ha prognosi più severa,
specialmente quando sono interessati il cuore, i polmoni o il tratto
gastroenterico, e in alcuni casi la disfunzione di questi organi può
essere letale18-21.
Diagnosi
e terapia
Le
immagini ecografiche, radiologiche e di RMN possono orientare nella
diagnosi e sono indispensabili nella valutazione dell'estensione
dei miofibromi, ma la diagnosi definitiva è istologica. Può
essere difficile differenziare istologicamente un miofibroma da un
fibrosarcoma, come è capitato a noi; in queste situazioni è
fondamentale l'ausilio della biologia molecolare per
l'identificazione del trascritto di fusione ETV6-NTRK322,23,
costitutivo del fibrosarcoma e assente nel miofibroma. Nel nostro
caso all'interno della stessa lesione si aveva un pattern
compatibile con miofibroma in certe aree e con fibrosarcoma in altre,
ma l'assenza della traslocazione specifica fa propendere per un
comportamento più simile a quello del miofibroma.
Prognosi
L'exeresi
chirurgica delle lesioni è risolutiva nell'80-90% dei casi;
tuttavia, data la tendenza alla regressione spontanea, è
possibile un atteggiamento di vigile attesa, limitando l'intervento
alle sole lesioni sintomatiche o che coinvolgano organi vitali.
Un
follow-up annuale è raccomandato per almeno tre anni dopo
l'escissione24. La comparsa di nuovi miofibromi è
stata riportata sia durante l'adolescenza, sia seppur meno
frequentemente, nell'età adulta25. L'impiego di
chemioterapia e radioterapia è controverso e riservato ai casi
più gravi; la riduzione e la scomparsa delle lesioni è
riportata in alcuni studi dopo somministrazione di Vincristina,
Adriamicina e Ciclofosfamide26. Attualmente il nostro
paziente ha 2 anni, esegue controlli periodici e non ha presentato
recidive, né nuove lesioni.
CONCLUSIONI
Il
riscontro di una lesione nodulare del capo richiede un percorso di
diagnosi differenziale che si avvale della diagnostica per immagini e
spesso della biopsia. L'escissione radicale è in molti casi
risolutiva.
- Li SL, Zhang XL, Han HX, et al. Imaging features of primary bone lymphoma and its histopathology. Nan Fang Yi Ke Da Xue Xue Bao 2007;27:201-4.
- Lieberman PH, Jones CR, Steinman RM, et al. Langerhans cell (eosinophilic) granulomatosis: a clinicopathologic study encompassing 50 years. Am J Surg Pathol 1996;20:519-52.
- Gibson SE, Prayson RA. Primary skull lesions in the pediatric population: a 25-year experience. Arch Pathol Lab Med 2007;131:761-6.
- Behrman RE, Kliegman RM, Jenson HB. Nelson. Textbook of Pediatrics. 17th editon. 2003, WB Saunders Company.
- Chung EB, Enzinger FM. Infantile myofibromatosis. Cancer 1981;48:1807-18.
- Lucas DR, Unni KK, McLeod RA, O'Connor MI, Sim FH. Osteoblastoma: clinicopathologic study of 306 cases. Hum Pathos 1994;25:11734.
- Cervoni L, Innocenzi G, Raguso M, Salvati M, Caruso R. Osteoblastoma of the calvaria: report of two cases diagnosed with MRI and clinical review. Neurosurg Rev 1997;20:5154.
- Barnes L. Solitary hemangioma of bone. In: Barnes L, ed. Surgical Pathology of the Head and Neck. New York: Marcel Dekker, Inc, 2001:110912.
- Adler CP, Wold L. Haemangioma and related lesions. In: Fletcher CD, Unni KK, Mertens F, eds. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002:3201. World Health Organization Classification of Tumours.
- Kaban LB, Mulliken JB. Vascular anomalies of the maxillofacial region. J Oral Maxillofac Surg 1986;44:20313.
- Williams JO, Schrum D. Congenital fibrosarcoma: Report of a case in a newborn infant. AMA Arch Pathol 1951;51:54852.
- Smith KJ, Skelton HG, Barrett TL, et al. Cutaneous myofibroma. Mod Path 1989;2:6039.
- Bracko M, Cindro L, Golouh R. Familial occurrence of infantile myofibromatosis. Cancer 1992;69:12949.
- Parker RK, Mallory SB, Baker GF. Infantile myofibromatosis. Pediatr Dermatol 1991;8:12932.
- Bellman B, Wooming G, Landsman L, et al. Infantile myofibromatosis: A case report. Pediatr Dermatol 1991;8:3069.
- Enzinger FM, Weiss SW. Fibrous tumors of infancy and childhood. In: Enzinger FM, Weiss SW, eds. Soft tissue tumors. London: CV Mosby, 1995:231-68.
- Coffin CM, Dehner LP. Congenital tumours. In: Stocker JT, Dehner LP, eds. Pediatric pathology. Philadelphia: JB Lippincott, 1992.
- Wiswell TE, Davis J, Cunningham BE, et al. Infantile myofibromatosis: the most common fibrous tumor of infancy. J Pediatr Surg 1988;23:314-8.
- Tamburrini G, Gessi M, Colosimo C Jr, et al. Infantile myofibromatosis of the central nervous system. Childs Nerv Syst 2003;19:650-54.
- Davies RS, Carty H, Pierro A. Infantile myofibromatosis: a review. Brit J Radiol 1994;67:619-23.
- Moore JB, Waldenmaier N, Potchen EJ. Congenital generalized fibromatosis: A new management strategy provided by magnetic resonance imaging. Am J Dis Child 1987;141:714-6.
- Argani P, Fritsch MK, Shuster AE, et al. Reduced sensitivity of paraffin-based RT-PCR assays for ETV6-NTRK3 fusion transcripts in morphologically defined infantile fibrosarcoma. Am J Surg Pathol 2001;25:1461-4.
- Argani P, Fritsch M, Kadkol SS, Schuster A, Beckwith JB, Perlman EJ. Detection of the ETV6-NTRK3 chimeric RNA of infantile fibrosarcoma/cellular congenital mesoblastic nephroma in paraffin-embedded tissue: application to challenging pediatric renal stromal tumors. Mod Pathol 2000;13:29-36.
- Stanford D, Rogers M. Dermatological presentations of infantile myofibromatosis: a review of 27 cases. Australas J Dermatol 2000;41:156-61.
- Jennings T, Duray PH, Collins FS, et al. Infantile myofibromatosis: evidence for an autosomal-dominant disorder. Am J Surg Pathol 1984;8:529-36.
- Raney B, Evans A, Granowetter L, et al. Non surgical management of children with recurrent or unresectable fibromatosis. Pediatrics 1987;79:394-8.
Vuoi citare questo contributo?