Gennaio 2005 - Volume VIII - numero 1
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Manifestazioni
ematologiche nelle malattie mitocondriali con mutazione del mt-DNA
Dipartimento
di Scienze Pediatriche dell'Università di Torino, Divisione
di Ematologia pediatrica
°
Istituto Nazionale “C. Besta” di Milano, Divisione di Genetica
molecolare
*Ospedale
Infantile Regina Margherita, Anatomia Patologica
Indirizzo
per corrispondenza:
rkyklos@mail2world.com
Keywords:Haematological manifestations, mitochondrial disease, Pearson
syndrome
Summary:We report a 10-month-old girl who, at the age of 4 months, was
admitted for increasing pallor. Severe normocytic anaemia,
neutropenia and thrombocytopenia and typical diffuse vacuolization of
marrow haemopoietic precursors were present. She also presented with
mild lactic acidosis. Molecular analysis of mt-DNA revealed an 8.000
bp single macrodeletion. Unexpectedly, anaemia was not sideroblastic
and there was no exocrine pancreatic dysfunction. Pearson
marrow/pancreas syndrome is a usually fatal mitochondrial disease
that involves the haematopoietic system, and usually also exocrine
pancreas, liver and kidneys. Other mitochondrial cytopathies with
mt-DNA mutation can have haematological manifestations, most of all
sideroblastic anaemia. Mitochondrial diseases should be considered in
the differential diagnosis of anaemia/pancytopenia in infancy.
Giulia ha
4 mesi, pesa 5.270 g (nata di 38 w. e di 2.380 g), presenta discrete
condizioni generali ma la madre riferisce inappetenza e soprattutto
pallore ingravescente nel corso delle ultime settimane. Il nostro
esame obiettivo conferma lo spiccato pallore, non evidenzia
malformazioni, epato-splenomegalia od altri elementi clinicamente
rilevanti. Le condizioni generali sono effettivamente discrete. Le
urine sono normocromiche e le feci normocoliche.
Decidiamo
di sottoporre la piccola ad alcuni esami ematochimici:
- L'esame emocromocitometrico rileva una anemia normocromica normocitica grave (con spiccata riduzione del numero di reticolociti rispetto alla norma) nell'ambito di una generale pancitopenia (lieve piastrinopenia, moderata neutropenia).
- L'osservazione delle emazie allo striscio e la valutazione dell'assetto marziale rendono improbabile una carenza marziale (non ipocromia o anisopoichilocitosi delle emazie, buona saturazione della transferrina)
- Non sono presenti segni di emolisi (bassa reticolocitosi, LDH, aptoglobina e bilirubina frazionata normali) neanche di natura autoimmune (test di Coombs negativo)
Vista
l'entità dell'anemia e l'assenza di una causa evidente
decidiamo di effettuare subito un agoaspirato midollare
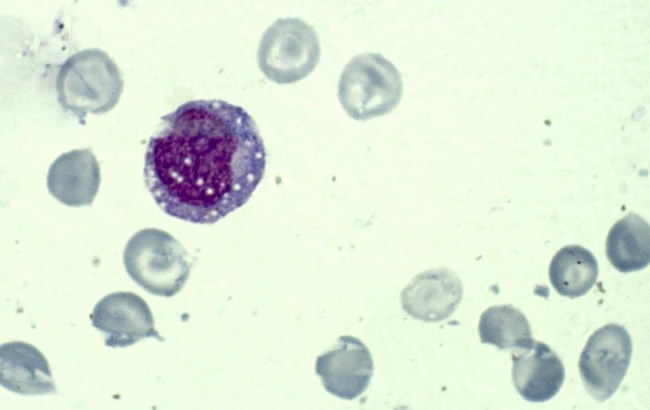
- L'analisi morfo-citologica mostra un quadro di cellularità ridotta con progenitori eritroidi molto rari (M/E 4/1) ma megacariociti presenti. Evidente anche una lieve displasia della serie mieloide. Risultano assenti cellule di appetto blastico. Numerosiprecursori midollari presentano significativamente unaspiccata vacuolizzazione. (vedi figura 1)
- Sempre su midollo valutiamo anche il cariotipo che risulta normale (46,XX), l'immunofenotipo non significativo ed escludiamo la presenza di virus con possibile effetto aplastizzante (PCR su midollo per parvovirus B19, HHV-6, \CMV ed EBV negative)
Effettuiamo
ulteriori esami ematici e ricerchiamo dei segni di malassorbimento:
- L'analisi del ciclo cellulare non evidenzia le alterazioni caratteristiche dell'anemia di Fanconi.
- Non sono evidenziabili grassi fecali ed il pancreas esocrino non mostra segni di ipofunzionalità
Sulla
base del quadro midollare andiamo a ricercare altri possibili segni
di malattia mitocondriale e li troviamo:
- All'E.G.A. è presente una lieve acidosi metabolica
- Il livello di lattato è lievemente aumentato
- Sono presenti delle alterazioni complesse dei livelli di acidi organici urinari
Inviamo
un campione di sangue all'Istituto “C. Besta” di Milano dove
viene riscontrata la presenza di una macrodelezione (8.000 bp)
del DNA mitocondriale (mt-DNA).
Il quadro
risulta quindi compatibile con una Sindrome di Pearson atipica
Sono
presenti alcuni degli elementi tipici della sindrome:
- La macrodelezione a carico del mt-DNA
- La vacuolizzazione dei precursori emopoietici
- L'anemia grave
Altri
dati sono compatibili con essa (la lieve acidosi e la modesta
iperlattacidemia, le alterazioni complesse degli acidi organici
urinari, la pancitopenia, le transaminasi mosse)
Tuttavia
alcuni elementi risultano atipici:
- A livello midollare paiono assenti i sideroblasti (anemia non sideroblastica)
- La funzionalità pancreatica risulta conservata
- Le condizioni generali sono soddisfacenti, le acquisizioni psico-motorie normali per l'età, la funzionalità renale normale.
- Origine: Probabilmente procarioti primitivi inglobati dalla cellula eucariotica primitiva 1.5 bilioni di anni fa. Questo evento ha portato ad una relazione simbiotica favorevole sempre più profonda tanto che oggi i mitocondri hanno perso la maggior parte del proprio genoma. Essi sono divenuti dipendenti dalle proteine codificate nel nucleo e si sono “specializzati” nella produzione di energia (catena ossidativa). (Teoria endo-simbiontica di Lynn Margulis,1981)
- Caratteristiche: Presenti in tutte le cellule in molti esemplari (fino ad alcune migliaia), contengono in 2-10 copie un proprio materiale genetico (mt-DNA) delle dimensioni di circa 16.500 bp codificante per 13 subunità della catena respiratoria (su circa 82 totali), 2 RNA ribosomiali e 22 RNA di trasferimento (non sono presenti introni). Meccanismi di riparazione del DNA deficitari con conseguente elevato tasso di mutazione. Funzione principale: produzione di energia (ciclo di Krebs, fosforilazione ossidativa, ossidazione degli acidi grassi). Altre funzioni note: sintesi dell'eme, eliminazione dell'ammonio, mediazione dell'apoptosi. Sono praticamente sempre di origine materna.
Le
malattie mitocondriali (“citopatie mitocondriali”) possono essere
ereditarie od acquisite, associate ad un difetto del DNA
mitocondriale oppure nucleare. Il difetto può essere
primitivamente a carico dei geni codificanti le subunità della
catena respiratoria o questa può essere coinvolta solo
secondariamente. Infine le mutazioni a carico del DNA mitocondriale
(mt-DNA) possono essere di vario tipo.
CLASSIFICAZIONE
DELLE CITOPATIE MITOCONDRIALI
- Associate a difetto dei geni codificanti per le subunità della catena respiratoria
- da mutazione mt-DNA
- da mutazione n-DNA
- Associate a difetto secondario della catena respiratoria
- da mutazione n-DNA
- da danno acquisito del metabolismo mitocondriale
MUTAZIONI
POSSIBILI A CARICO DEL mt-DNA
- riarrangiamenti
- micro/macro delezioni
- duplicazioni
- mutazioni a carico del t-RNA
- mutazioni puntiformi
- mutazioni a carico del r-RNA
Caratteristiche
peculiari delle patologie con difetto del mt-DNA sono la cosiddetta
“eteroplasmia” ed il conseguente “effetto
soglia”. All'interno delle diverse cellule, tessuti ed organi
degli individui affetti è presente un mix di molecole normali
e mutanti di mt-DNA che è alla base della estrema variabilità
individuale del fenotipo.
LaSindrome di Pearson ma anche la Sindrome di Kearns-Sayre e la
Oftalmoplegia Esterna Progressiva sono associate a macrodelezioni
del mt-DNA e sono normalmente sporadiche. Anche la
Sindrome di Wolfram può essere associata a difetto del mt-DNA.
E' una
citopatia mitocondriale associata a macrodelezione del mt-DNA ad
espressione prevalentemente ematologica ma con distribuzione
ubiquitaria del mt-DNA mutante. Tipica della sindrome è anche
una compromissione più o meno rilevante della funzionalità
del pancreas esocrino. Costante l'anemia normo-macrocitica
grave (generalmente sideroblastica), frequenti la neutropenia e la
piastrinopenia. I progenitori midollari sono intensamente
vacuolizzati.
Clinical
Synopsis (da OMIM-Online Mendelian Inheritance in Man-John
Hopkin's University)
- Heme : Refractory sideroblastic anemia. Vacuolization of marrow precursors.
- GI : Exocrine pancreatic dysfunction. Malabsorption. Pancreatic fibrosis. Splenic atrophy.
- Growth : Low birth weight. Failure to thrive.
- Metabolic : Insulin-dependent diabetes mellitus. Metabolic acidosis. Lactic acidosis.
- Misc : Frequently death in infancy.
- Lab : 3-methylglutaconicaciduria. Complex organic aciduria.Mitochondrial deletions. Renal Fanconi syndrome. Increased ketone body or lactate/pyruvate plasma ratios.
- Inheritance : Mitochondrial.
Storia
Naturale
La
Sindrome di Pearson esordisce nel 70% dei casi entro i sei mesi di
vita, è frequentemente fatale ed il decesso avviene più
comunemente nei primi anni di vita o comunque in età
pediatrica. Le più frequenti cause di morte sono di tipo
infettivo (sepsi batterica secondaria a neutropenia ed atrofia
splenica) o legate all'acidosi (crisi metabolica) o
all'insufficienza epatica. Nei pochi pazienti che sopravvivono più
a lungo si osserva in genere un miglioramento spontaneo del quadro
ematologico e in alcuni casi si assiste ad uno “shift” del
fenotipo che da prevalentemente ematologico diventa miopatico od
encefalopatico. Una parte degli affetti sviluppa la Sindrome di
Kearns-Sayre. La terapia è generalmente di supporto
(emotrasfusioni, complessi multivitaminici). Segnalati casi
sottoposti con successo ad allo-TMO.
Ladiagnosi differenziale si pone principalmente con:
- altre cause di citopenia congenita (an. di Diamond-Blackfan, an. di Fanconi, s. Shwachmann)
- altre cause di anemia sideroblastica (anemia tiamino-sensibile)
- altre cause di vacuolizzazione progenitori emopoietici (deficit di fenilalanina, riboflavina o rame, intossicazione da CAF o alcool)
- altre cause di mitocondriopatia
- altre mitocondriopatie associate a macrodelezione mt-DNA (Sindrome di Kearns-Sayre, Oftalmoplegia esterna progressiva)
Altra
mitocondriopatia normalmente associata a macrodelezioni (spesso
identiche a quelle del Pearson e della Oftalmoplegia Esterna
Progressiva*) caratterizzata principalmente da una oftalmoplegia
esterna lentamente progressiva, una retinopatia con deposito
di pigmento e disturbi a carico della conduzione cardiaca (Kearns and
Sayre, 1958). Ci sono casi non associati a mutazione Del mt-DNA.
Spesso è presente una anemia sideroblastica.
Caratterizzata
principalmente dall'oftalmoplegia (con coinvolgimento
dell'elevatore della palpebra, dell'orbicolare e dei muscoli
estrinseci dell'occhio) con perdita progressiva della motilità
oculare. Anch'essa si può associare ad anemia
sideroblastica.
Caratterizzata
da diabete insipido, diabete mellito, atrofia ottica e sordità
(DIDMOAD). Elementi fondamentali sono il diabete mellito e l'atrofia
ottica. Spesso presenti altri sintomi neurologici, psichiatrici ed
endocrinologici. Alcuni casi sono autosomici, altri associati a
mutazioni del mt-DNA. Possono essere presenti: anemia
megaloblastica e/o sideroblastica, neutropenia epiastrinopenia.
DUBBI
ED IPOTESI
Pearson,
K-S e OEP: diverse facce della stessa medaglia?
Identiche
delezioni sono state segnalate nella S. di Pearson, nella S. di K-S,
e nella OEP. Nelle tre sindromi la distribuzione tissutale del mt-DNA
mutante è differente. In quella di Pearson ci sono elevati
livelli di mt-DNA mutante in tutti i tessuti ed in particolare nel
midollo. Nella Kearn-Sayres esso è presente ad alti livelli
soprattutto nei muscoli e nel sistema nervoso centrale. Nella
Oftalmoplegia la distribuzione del mt-DNA mutante è
probabilmente ancora più limitata.
Quale
il vero ruolo eziopatogenetico del difetto del mt-DNA?
Vari
elementi hanno indotto i ricercatori a chiedersi se le mutazioni del
DNA mitocondriale siano davvero alla base delle rispettive malattie.
Per alcune citopatie mitocondriali associate a difetti del mt-DNA
(spt. OEP e S.W.) più o meno recentemente è stata
ipotizzato che questi siano in realtà secondari ad un
primitivo difetto del n-DNA che fungerebbe da fattore predisponente
allo sviluppo di mutazioni a carico del genoma mitocondriale.
Una
mitocondriopatia nella patogenesi di tutte le anemie
sideroblastiche?
Una
mitocondriopatia (congenita o, più spesso, post-natale) con
conseguente alterazione della produzione di energia cellulare (e
quindi con difettoso metabolismo del ferro) potrebbe essere alla base
di un'anomala deposizione di ferro a livello degli eritroblasti
(anemia sideroblastica).
DA
RICORDARE:
-Un'anemia
normo-macrocitica (o anche una pancitopenia) nel lattante può
rappresentare l'esordio di una Sindrome di Pearson che quindi va
considerata nella diagnosi differenziale
-In
generale il quadro clinico di alcune malattie mitocondriali può
comprendere delle manifestazioni ematologiche (anemia, neutropenia,
piastrinopenia, vacuolizzazione dei progenitori)
- OMIM: Pearson marrow-pancreas syndrom
- De Lonlay P et al.: Manifestations hematologiques dans les erreurs innes du metabolisme. Arch Pediatr. 2002; 8: 822-35
- Thorburn DR et al.: Mitochondrial Disorders: Genetics, Counseling, Prenatal Dagnosis and Reproductive Options. Am J Med Gen. 2001; 106: 102-14
Vuoi citare questo contributo?