Rivista di formazione e aggiornamento di pediatri e medici operanti sul territorio e in ospedale. Fondata nel 1982, in collaborazione con l'Associazione Culturale Pediatri.
Ottobre 1999 - Volume II - numero 8
M&B Pagine Elettroniche
Pediatria per l'ospedale
Leβ-talassemie
Le
b-talassemie hanno rappresentato per la pediatria di qualche decina
di anni fa, e in parte lo sono ancora, uno dei più importanti
problemi di patologia.
Noi
pediatri italiani abbiamo il grande rimpianto di non aver
riconosciuto per primi la malattia e di aver lasciato che il dottor
Thomas Cooley nel 1925 la descrivesse in bambini di origine italiana,
portatori di una forte splenomegalia e di lesioni ossee. I nostri
vecchi pediatri, di fronte a casi del genere, e ce ne dovevano essere
a centinaia, se non a migliaia in alcune Regioni d'Italia, pensavano
alla sifilide, al morbo di Banti, alle anemie splenomegaliche
aspecifiche, all'ittero di Rietti-Greppi-Micheli, ma si lasciavano
sfuggire l'essenza del problema, perché non riflettevano a
sufficienza sui dati epidemiologici, sull'ereditarietà e
sull'individualità del quadro clinico. Sono stati necessari 50
e più anni perché i pediatri italiani si
riappropriassero di questa patologia (soprattutto Cao, 1988; Cao et
al., 1998) e tracciassero la strada che ha portato al trapianto di
midollo osseo (Lucarelli et al, 1997; Angelucci et al, 1997).
Epidemiologia
La
talassemia (T) è una malattia su base ereditaria, di tipo
autosomico recessivo, diffusa in tutto il mondo, e, per quanto ci
riguarda, concentrata soprattutto sulle coste del Mediterraneo (nelle
Regioni come la Sicilia, la Calabria, la Sardegna e l'Emilia
Romagna). Per le forti trasmigrazioni interne, oggi essa è
diffusa praticamente in tutte le parti d'Italia.
La
frequenza del gene patologico è molto elevata e può
essere presente in qualche area anche nel 10% della popolazione:
complessivamente in Italia viene calcolato che ci siano 2 milioni di
eterozigoti. Viene oggi ritenuto che la diffusione della T sia legata
al vantaggio che l'eterozigote presenta nei confronti della malaria
da Plasmodium falciparum; la selezione naturale attraverso
decine di migliaia di anni ha portato all'attuale distribuzione
(Pattanapanyasat et al., 1999). Nelle diverse popolazioni la T ha
avuto un'origine indipendente, come è dimostrato dalle
numerose diverse mutazioni, caratteristiche di ogni popolazione.
Patologia
molecolare
I geni
che regolano la sintesi della b-globina sono tutti sul braccio corto
del cromosoma 11 e sono formati da circa 60.000 basi di nucleotidi.
La b-talassemia si manifesta clinicamente, nel corso del primo anno
di vita, quando si riduce la sintesi dell'emoglobina fetale (HbF),
senza che l'HbF venga rimpiazzata, come di norma, dall'emoglobina
A1.
Per la
b-talassemia sono state riscontrate oltre 200 mutazioni, per la gran
parte piccole sostituzioni di nucleotidi e solo talvolta di
delezioni.
Qualunque
sia il tipo di mutazione, le conseguenze possono essere
fondamentalmente di due tipi:
- non c'è
per niente sintesi della catena della b-globina (b0-talassemia) o
-esiste
una riduzione della sintesi (b+-talassemia)
Il quadro
clinico è strettamente correlato all'una o all'altra
situazione.
Cause
dell'anemia
Nelle
b-talassemie non trattate, l'eritropoiesi può aumentare anche
di 30 volte in confronto al normale, ma in più del 95% di essa
si tratta di un'eritropoiesi inefficace, cioè della morte del
globulo rosso, quando ancora non ha lasciato il midollo osseo. La
causa dell'inefficacia dell'eritropoiesi risiede in un numero elevato
di effetti dannosi, dovuti all'eccesso di catene globiniche a, che
rimangono libere nel globulo rosso, non potendosi unire alle catene
b, che sono mancanti o in numero ridotto. La splenomegalia,
conseguente all'aumento dell'attività emocateretica della
milza, contribuisce all'aggravamento dell'anemia. L'attività
eritropoietica, sotto lo stimolo dell'aumentata secrezione di
eritropoietina, sintetizzata dal rene in risposta all'anemia, è
enormemente accentuata ed è la responsabile dei focolai di
eritropoiesi extramidollare nel torace, nella regione paraspinale e
nei parenchimi; l'eritropoiesi extramidollare è d'altra parte
responsabile della presenza di eritroblasti nel sangue circolante,
perché la soglia, che ne impedisce il passaggio in circolo, è
presente solo nel midollo osseo e non a livello dei focolai di
eritropoiesi extramidollare.
L'espansione
del midollo, a livello delle ossa, porta d'altra parte alle deformità
del cranio e della faccia, all'osteopenia e ai difetti focali di
mineralizzazione (microfratture e osteomalacia). Infine l'iperplasia
midollare porta a un aumento del riassorbimento di ferro e a una
progressiva deposizione di ferro nei tessuti.
Fig
1:Effetti dell'eccesso di produzione delle catene a della globina
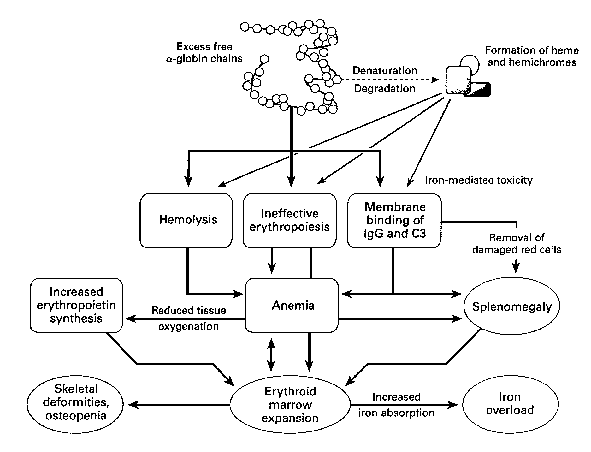
L'eccesso
di catene globiniche a e dei loro prodotti di degradazione porta alla
formazione di precipitati all'interno dei precursori del globulo
rosso, con conseguenti difetti di maturazione ed eritropoiesi
inefficace. All'anemia contribuisce l'emolisi, dovuta alla presenza
d'inclusioni nei globuli rossi e alla sofferenza delle loro membrane,
in seguito all'eccesso di catene globiniche a, aggregate fra loro, e
ai loro prodotti di degradazione. L'anemia stimola la sintesi di
eritropoietina con conseguente intensa proliferazione inefficace del
midollo, che a sua volta determina deformità scheletriche e
una grande varietà di alterazioni morfologiche e metaboliche.
L'anemia è ulteriormente aumentata dall'emodiluizione, causata
dal passaggio del sangue attraverso il midollo osseo espanso e
attraverso la milza, aumentata di volume in seguito
all'intrappolamento di enormi quantità di globuli rossi
deformati. L'espansione del midollo osseo è alla base delle
caratteristiche deformità del cranio e del volto, della grave
osteopenia e dell'aumentato assorbimento di ferro.
(da
Olivieri, 1999).
Accanto
all'eritropoiesi inefficace, un'altra importante causa di anemia
è l'indeformabilità dei globuli rossi, caratteristica
della b- Talassemia (Dondorp et al, 1999).
L'eterogeneità
dei globuli rossi e la produzione di emoglobina fetale
Come
abbiamo visto l'emoglobina fetale diminuisce progressivamente dopo
la nascita, anche se una parte di essa continua a essere
sintetizzata: questa ridotta quantità, presente in quantità
diversa da un globulo rosso all'altro, non è affatto
sufficiente per compensare la ridotta sintesi di HbA1.La sintesi di
HbF può in qualche caso raggiungere i 2-4 g/dL a tutto
vantaggio del piccolo paziente. Le diverse forme dei globuli rossi,
caratteristiche degli strisci dei bambini con talassemia major
dipendono proprio dalla diversa quantità di HbF in essi
contenuta, ma soprattutto dall'eccessiva quantità di catene
globiniche a, spesso aggregate fra loro in tetrameri. L'eccessiva
concentrazione di catene a e dei loro prodotti di degradazione
portano a modificazioni della membrana e dello scheletro dei globuli
rossi: la stessa struttura della spectrina risulta
profondamente modificata.
Per far
comprendere quanto alcuni pediatri italiani fossero lontani dalla
giusta interpretazione dei meccanismi regolatori della sintesi
dell'HbF, ricordo che una quarantina di anni fa vennero pubblicati,
su una rivista pediatrica italiana, alcuni lavori sull'effetto delle
radiazioni del midollo osseo in bambini con talassemia major,
praticate allo scopo di abbassare la produzione di HbF, come se fosse
questa la responsabile del quadro clinico da essi mostrato.
Forme
cliniche
Oggi in
seno alla b-talassemia si riconoscono 4 forme cliniche, di gravità
crescente:
1)stato di portatore silente
2)tratto talassemico, caratterizzato da lieve anemia, ridotto
volume cellulare medio, ridotta concentrazione media di Hb per
cellula, aumentate concentrazioni di HbA2 (oltre il 3,5%). Questa
forma e la precedente non si accompagnano ad alcun rilievo clinico,
per cui sono spesso rilievi casuali nel corso di esami di
laboratorio, eseguiti per altro scopo
3)talassemia intermedia, giunge all'attenzione del medico
tardivamente e richiede solo di rado l'uso delle trasfusioni.
4)talassemia major (o morbo di Cooley), giunge
all'attenzione del medico precocemente, per lo più nel primo
anno di vita: in seguito questi pazienti sopravvivono solo grazie a
numerose trasfusioni di sangue.
Il
fenotipo può essere diverso a seconda del genotipo: per
esempio una doppia ereditarietà per a e b-talassemia può
ridurre le conseguenze dello sbilanciamento delle rispettive globine.
Risultato analogo si verifica nella micro-drepanocitosi, quando sia
presente b-talassemia lieve e presenza di emoglobina S.
Complicazioni
della malattia
Le
conseguenze della b-talassemia allo stato omozigote sono
molteplici e tutte di estrema importanza per il soggetto che ne sia
affetto.
Di
recente è stata sempre più spesso rilevata la presenza
di deficit neurologici in bambini con b-talassemia, dovuti a
alla presenza di uno stroke emorragico cerebrale per fenomeni
tromboembolici (Borgna-Pignatti et al., 1998; Incorpora et al.,
1999).
Il
sovraccarico di ferro dei tessuti rimane tuttavia la più
importante complicazione della b-talassemia e rappresenta il
principale obiettivo del trattamento.
Nei
pazienti che non ricevono trasfusioni, l'aumento del ferro tissutale
varia fra 2 e 5 g per anno, a seconda dell'espansione del tessuto
eritroide. Ma se un paziente riceve regolari trasfusioni, al ritmo di
una ogni 15-20 giorni, questa quantità può
raddoppiarsi. L'accumulo di ferro è dovuto alla caratteristica
del nostro organismo di trattenere ferro a qualsiasi costo: nel
normale infatti il ferro non viene eliminato in quantità
superiori a 1 mg al giorno, tanto quanto se ne assorbe. Ma se
l'assorbimento è elevato, in seguito all'anemia, e se il ferro
è introdotto nell'organismo con le trasfusioni, senza essere
adeguatamente utilizzato per la sintesi di emoglobina, esso si
accumula progressivamente nei vari tessuti.
Gli
effetti tossici dell'accumulo del ferro, in mancanza di un'adeguata
chelazione, già sono presenti nella prima decade di vita. Nel
nostro organismo abbiamo una proteina (la transferrina), la cui
funzione principale è il trasporto del ferro: quando il ferro
è talmente abbondante da superare le sue capacità di
trasporto, esso si accumula progressivamente in circolo, non legato
alla transferrina, fino a promuove la creazione di radicali liberi,
che determinano effetti tossici a vari livelli. Sebbene il nostro
organismo abbia molti sistemi enzimatici ad azione antiossidante
(superossido-dismutasi, catalasi, glutatione perossidasi), quando il
carico di ferro sia eccessivo, essi non sono più sufficienti a
impedire il danno ossidativo, legato a questi prodotti.
In
assenza di un'appropriata terapia chelante, l'accumulo di ferro è
il responsabile della sofferenza progressiva e quindi della
disfunzione di alcuni parenchimi: il primo luogo del cuore, ma anche
del fegato e delle ghiandole endocrine. A carico del cuore si
manifesta prima ipertrofia e poi dilatazione, con degenerazione delle
fibre miocardiche e talvolta fibrosi. In soggetti non sottoposti a
terapia chelante, ma trattati regolarmente con trasfusioni, quadri
clinici di sofferenza cardiaca si manifestano dopo circa 10 anni
dall'inizio della terapia trasfusionale e possono essere aggravati
dalla miocardite e dall'ipertensione polmonare. La sopravvivenza dei
pazienti con b-talassemia è determinata dall'entità
del sovraccarico di ferro nel cuore.
Un'altra
possibilità di morte viene per questi pazienti anche dal
sovraccarico di ferro del fegato, aggravato spesso dalla
sovrapposizione di un'epatite B o C (Prati et al., 1998), in
conseguenza delle numerose trasfusioni di sangue, alle quali vengono
regolarmente sottoposti questi pazienti. Basta che siano passati due
anni dall'inizio delle trasfusioni perché insorgano, a carico
del fegato, un'eccessiva formazione di collageno e fibrosi portale.
In assenza di terapia chelante la cirrosi si può sviluppare
alla fine della prima decade di vita. Il rischio di danno epatico è
aumentato quando la concentrazione di ferro superi i 7 mg per grammo
di tessuto epatico, allo stato secco.
Sulle
conseguenze del sovraccarico di ferro nelle ghiandole
endocrine si sono soffermati, negli ultimi 10 anni, soprattutto
ricercatori italiani (Italian Working Group, 1995, Caruso-Nicoletti
et al., 1998). Le complicazioni endocrine sono più frequenti
nei soggetti con talassemia major di maggiore età: ritardata
maturazione sessuale (presente nel 50% dei soggetti, sia di sesso
femminile che maschile), amenorrea secondaria (presente in circa un
quarto dei soggetti di sesso femminile in età superiore ai 15
anni), diabete mellito (in circa il 5% degli adulti), sofferenze
tiroidee, paratiroidee e surrenaliche.
l
sovraccarico di ferro può alla lunga determinare ipertensione
polmonare, dilatazione del ventricolo destro e malattia polmonare in
senso restrittivo.
Prevenzione
e trattamento
I
programmi di screening, la prevenzione e la diagnosi prenatale hanno
portato a una decisa riduzione della nascita di bambini con
talassemia major, almeno nell'area del Mediterraneo (Cao et al.,
1998). Gli screening si basano sull'abbassamento degli indici del
volume cellulare medio (da ricordare che l'abbassamento dei valori
del CMV si ritrova anche nell'anemia ferropriva, nella talassemia ? e
nell'avvelenamento cronico da piombo), del contenuto medio della
concentrazione di Hb per globulo rosso e infine nell'aumento della
concentrazione di HbA2: la percentuale di HbA2 (>3,5%) è
aumentata anche in presenza di deficienza di ferro (Madan et al.,
1998). La diagnosi prenatale, inizialmente basata sui campioni di
sangue fetale e sulla valutazione della sintesi delle catene
emoglobiniche nel sangue fetale, è oggi soprattutto eseguita
sull'esame del DNA fetale, ottenuto dai campioni di villi coriali.
Le
trasfusioni di globuli rossi
La
decisione di quando iniziare il trattamento cronico trasfusionale è
sempre abbastanza difficile, perché non esiste un unico
parametro di valutazione: essa si basa essenzialmente sulla presenza
e la gravità dei sintomi e dei segni di anemia, compreso il
ritardo di sviluppo e di crescita.
Gli
scopi della trasfusione sono molteplici:
-
correggere l'anemia
-
sopprimere l'eccesso di eritropoiesi e limitarne le conseguenze
- inibire
l'aumento dell'assorbimento del ferro, legato all'anemia
Fino
a una decina di anni fa, la tendenza era verso "l'ipertrasfusione",
che purtroppo si associava a un aumento del sovraccarico di ferro.
Oggi la tendenza generale di tutti i centri è di non superare
i 9,5 g/dL di Hb: questi livelli comportano un'adeguata soppressione
dell'attività del midollo osseo e hanno una relativamente
bassa percentuale di accumulo di ferro.
La
deferoxamina
L'impiego
della deferoxamina (D), l'unico agente chelante del ferro in
commercio in Italia, è risultato decisivo per la prognosi dei
pazienti con talassemia major. E' stato osservato che l'impiego di
dosi adeguate di deferoxamina prevengono la morte precoce per
insufficienza cardiaca, perché permettono di mantenere la
concentrazione di ferro nel fegato al di sotto dei 15 mg/g di fegato
allo stato secco, livello al di sotto del quale è notevolmente
ridotto il rischio di malattia clinica. La D, secondo i moderni
regimi d'impiego, rende vicine al normale le concentrazioni di ferro
del fegato e arresta la progressione della fibrosi epatica verso la
cirrosi.
Una
riduzione del sovraccarico di ferro, ha effetti favorevoli anche
sulla funzionalità delle ghiandole endocrine e quindi
indirettamente sulla maturazione sessuale. La D previene anche il
diabete mellito, ma non ha alcun effetto quando questo si sia già
instaurato.
Nella
pratica clinica fino a oggi la valutazione del sovraccarico di ferro
veniva fatto sulla base della determinazione della ferritina sierica,
ma chi si è interessato di questo argomento sa quanto siano
fallaci i dati della ferritinemia e come questi siano influenzabili
dalla funzionalità del fegato e da altri fattori. Per avere
un'idea precisa della concentrazione di ferro nei tessuti è
necessario invece valutarne i livelli sopra un frammento di tessuto
epatico, ottenibile con una biopsia.
Una
metodica molto meno invasiva,.e altrettanto valida, è la
determinazione della concentrazione di ferro nel fegato, mediante lasusceptometria magnetica, disponibile al momento attuale sono
in due centri USA. La più comune tecnica della RM non si è
dimostrata utile per la determinazione del ferro epatico.
Il
trapianto di midollo osseo
Il
trapianto di midollo osseo da donatori HLA-identici è stato
eseguito in tutto il mondo in oltre 1.000 pazienti con grave
b-talassemia (Giardini, 1997). I bambini, che non presentino fattori
di rischio, come una grave epatomegalia, una fibrosi portale e
l'inefficacia della terapia chelante, hanno una probabilità di
successo di oltre il 90%, almeno nei 3 anni dopo il trapianto. In
quelli che hanno i 3 fattori di rischio sopra ricordati (si tratta
quasi esclusivamente di adulti), il successo raggiunge ugualmente il
60%.
L'eritropoietina
L'uso
dell'eritropoietina nella talassemia major si accompagna a
un'aumentata sintesi di catene ? e quindi di emoglobina F: Il
fattore limitante è rappresentato dall'alto costo
(Rachmilewitz et al., 1998).
Terapie
sperimentali
Qualche
anno fa venne proposto un nuovo chelante, che aveva il grande
vantaggio di essere somministrabile per bocca, il deferiprone.
Purtroppo le attesa vennero deluse, perché il deferiprone non
ha dimostrato di controllare adeguatamente i depositi di ferro e
inoltre perché esso può determinare o aggravare la
fibrosi epatica (Olivieri et al., 1998).
Un'altra
strada, seguita per migliorare la prognosi dei pazienti con
talassemia major è quella di potenziare la sintesi
dell'HbF (proprio il contrario di quello che faceva quel pediatra
di una quarantina di anni fa): anche in questo caso alle prime
speranze hanno seguito profonde delusioni.
Anche laterapia genica non ha portato per ora ad alcun successo
pratico.
Bibliografia
Angelucci
E, Giardini , Brittenham GM, Lucarelli G - Hepatic iron concentration
and body iron stores determined by quantitive phlebotomy in patients
cured of thalassemia major by bone marrow transplantation -Blood 90, Suppl1, 265a, 1997
Borgna-Pignatti
C, Carnelli V, Caruso V et al Thromboembolic events in
beta-thalassemia major: an Italian multicenter study Acta
Haematol 99, 76-9, 1998
Cao A -
"Diagnosis of beta-talassemia intermedia at presentation".
In S. Fucharoen, Thalassemia: pathophysiology and management. Part B,
Vol. 23, New York, AL Liss 219-26, 1988
Cao A,
Galanello R, Rosatelli MC - Prenatal diagnosis and screening of the
hemoglobinopathies. Ballieres Clin Haematol11, 215-38, 1998
Caruso-Nicoletti
M, De Sanctis V, Capra M et al Short stature and body proportion
in thalassaemia J Pediatr Endocrinol Metab 11, Suppl 3,
811-6, 1998
Dondorp
AM, Chotivanich KT, Fucharoen S et al Red cell deformability,
splenic function and anemia in thalassemia Br J Haematol105, 505-8, 1999
Giardini
C - Treatment of beta-thalassemia - Curr Opin Hematol 4,
79-87, 1997
Italian
Working Group on Endocrine Complications in Non-endocrine Diseases.
Multicentre study on prevalence of endocrine complications in
thalassemia major - Clin Endocrinol 42, 581-6, 1995
Incorpora
G, Di Gregorio F, Romeo MA et alò Focal neurological
deficits in children with beta-talasemia major Neuropediatrics30, 45-8, 1999
Lucarelli
G, Giardini C, Angelucci E - Bone marrowe transplantation in
thalassemia, in JN Winter, Bllod steam cell transplantation,
Boston, Kluwer Academic 1997, 305-15
Madan N,
Sikka M, Sharma S, Rusia U Phenotypic expression of hemoglobin A2
in beta-thalassemia trait with iron deficiency Ann Hematol77, 93-6, 1998
Olivieri
NF, Brittenham GM, McLaren CE et al Long-term safety and
effectiveness of iron-chelation therapy with deferiprone for
thalassemia major N Engl J Med 339, 417-23, 1998
Olivieri
NF - The b-talassemias - N Engl J Med 341, 99-109, 1999
Pattanapanyasat
K, Yongvanitchit K, Tongtawe P et al Impairment of Plasmodium
falciparum growth in thalassemic red blood cells: further evidence by
using biotin labeling and flow cytometry - Blood 93,
3116-9, 1999
Prati D,
Zanella A, Farma E et al A multicentric prospective study on the
risk of acquiring liver disease in anti-hepatitis C virus negative
patients affected from homozygous beta-thalassemia Blood
92, 34670-4, 1998
Rachmilevitz
EA, Aker M The role of recombinant human erythropoietin in the
treatment of thalassemia Ann N Y Acad Sc 850, 129-38, 1998
Vuoi citare questo contributo?