Dicembre 2001 - Volume IV - numero 10
M&B Pagine Elettroniche
Contributi Originali - Casi contributivi
Miofibromatosi
congenita: un caso clinico
U.O. di
Pediatria, Campobasso
*U.O. di
Dermatologia, Rimini
**U.O. di
Dermatologia, Cesena
Introduzione
Nel
gruppo delle miofibromatosi congenite, a prescindere dalle
definizioni classificative fino ad ora proposte dai vari autori, si
riconoscono sostanzialmente quadri caratterizzati da esclusivo
interessamento cutaneo e quadri in cui le lesioni cutanee si
accompagnano a lesioni ossee, muscolari e viscerali. Mentre le forme
con impegno viscerale sono generalmente caratterizzate da una
prognosi grave quelle ad esclusivo interessamento cutaneo presentano
solitamente un decorso favorevole.
Le
lesioni cutanee della miofibromatosi congenita - al pari di altre
lesioni dell'età neonatale (emangiomi, mastocitomi,
istiocitosi a cellule di Langerhans) - dopo aver esaurito le iniziali
potenzialità proliferative subiscono invariabilmente una
spontanea regressione nei mesi successivi alla nascita.
Caso
clinico
Davide M.
nasce a termine da parto eutocico. L'anamnesi familiare è
negativa. Alla nascita si apprezzano alcuni elementi nodulari a sede
sottocutanea, non aderenti ai piani sopra e sottostanti e ricoperti
da cute apparentemente indenne. Le dimensioni sono variabili, da
pochi millimetri a circa due centimetri, e la consistenza è
duro-elastica. I distretti anatomici interessati sono il vertice, la
regione parietale sinistra, i glutei e il dorso. In seconda giornata
si registra la comparsa di un nuovo nodulo alla coscia sinistra e
successivamente di numerosi altri elementi al tronco, mentre le
lesioni del vertice e del dorso mutano le loro caratteristiche
morfologiche divenendo la prima più voluminosa (Fig.1), con
superficie mammellonata e la seconda francamente esofitica e di
colorito rosso violaceo (Fig.2).
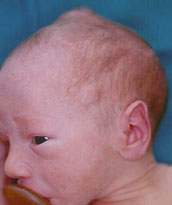 | 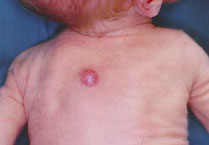 |
FIG.1 | FIG.2 |
Le
indagini laboratoristiche non presentano alterazioni significative,
TAC, ecografia addominale ed ecocardiogramma risultano nella norma.
L'esame radiologico dello scheletro evidenzia invece a livello della
teca cranica una spiccata deformazione in corrispondenza della
voluminosa lesione nodulare cutanea (Fig.3).
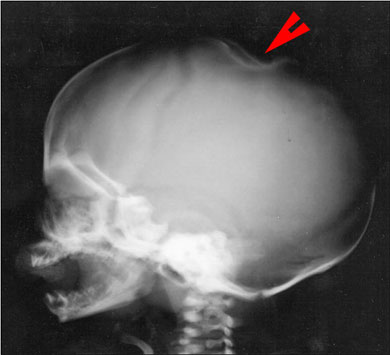 | |
FIG.3 | |
Una prima
biopsia cutanea (a livello della lesione nodulare del dorso) all'età
di 14 giorni pone in evidenza una struttura compatta costituita da
elementi cellulari fusiformi e collagene disposti in fasci tra loro
variamente intrecciati, suggerendo così la diagnosi di
miofibromatosi.
Nei mesi
successivi alla nascita nuovi elementi insorgono mentre altri
subiscono una spontanea involuzione. All'età di otto mesi
quasi tutti le lesioni sono regredite, ad eccezione di quelle del
capo e dell'addome (Fig.4).
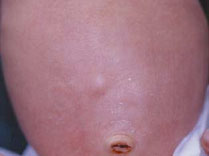 | |
FIG.4 | |
In
considerazione della loro prolungata persistenza, poco gradita ai
familiari, le due lesioni del capo (vertice e regione parietale
sinistra) vengono asportate chirurgicamente con buon esito
cicatriziale. In sede chirurgica si apprezza in entrambe le
localizzazioni una depressione macroscopica della teca cranica che,
come una nicchia, offre alloggio alle proliferazioni
miofibroblastiche. Le ulteriori indagini microscopiche
(immunoistochimica e microscopia elettronica) consentono di
convalidare la diagnosi clinica di miofibromatosi congenita.
Attualmente,
all'età di tre anni e mezzo, tutte le lesioni sono scomparse e
il bambino si presenta in buona salute
Commento
Il caso
presentato, che può essere certamente ascritto alla
miofibromatosi congenita ad elementi multipli con esclusivo
interessamento cutaneo, si caratterizza per la natura secondaria
della lesione ossea radiologicamente documentata a carico della teca
cranica (Fig.3). Come spesso accade il progressivo accrescimento di
una lesione cutanea in forma nodulare e di consistenza dura o
teso-elastica (sia essa un miofibroma o una lesione proliferativa di
altra natura, una cisti dermoide malformativa o una semplice cisti
sebacea) determina facilmente, in età pre e neonatale, una
depressione corrispondente delle strutture ossee sottostanti, specie
a livello del distretto cefalico.
Il caso
di Davide inoltre, nel confermare l'evoluzione autorisolutiva delle
lesioni cutanee, ne evidenzia la variabilità di presentazione
clinico-morfologica, non solo in termini di dimensioni e consistenza.
Infatti, accanto a lesioni nodulari endofitiche, mobili rispetto al
piano cutaneo e ricoperte da cute normale (Fig.4), coesistono lesioni
a sviluppo esofitico, coerenti con il piano cutaneo, che si presenta
ora di colorito normale (Fig.1) ora rosso-violaceo (Fig.2).
Infine,
vorremmo sottolineare che l'asportazione chirurgica delle lesioni non
è strettamente necessaria, poiché nella maggior parte
dei casi ai fini diagnostici può risultare sufficiente una
semplice biopsia punch e poiché le lesioni sono destinate a
regressione spontanea. Nel nostro caso, tuttavia, si è
ritenuto opportuno procedere all'exeresi radicale delle lesioni del
capo all'età di 8 mesi per la loro spiccata propensione
all'accrescimento, a dispetto della tendenza involutiva dimostrata
dalla gran parte delle altre lesioni, per l'espresso desiderio dei
familiari e, non ultimo, per poter disporre di maggiori quantità
di tessuto da destinare allo studio istopatologico. La possibilità
di intervenire in regime di day hospital, senza esami preliminari e
con anestesia combinata (anestesia locale + breve sedazione
generale), ci ha certamente facilitato nell'assumere questa
decisione.
Biobliografia
1 -
Bonifazi E, Filotico R, Mazzotta F, Ruggiero G, Modesti A.
Miofibromatosi congenita generalizzata con interessamento
neurologico. Eur J Pediat Dermatol 1991;1:141-50
2 -
Schrodt BJ, Callen JP. A case of congenital multiple myofibromatosis
developing in an infant. Pediatrics 1999;104:113-5
3 -
Giannakopoulou C, Hatzidaki E, Giannakopoulos K, Matalliotakis I,
Koumantakis E, Kalmanti M. Infantile myofibromatosis: a case study
and review of literature. J Dermatol 1999;26:595-8
4 -
Dimson OG, Drolet BA, Southern JF, Rock A, Winthrop AL, Esterly NB.
Congenital generalized myofibromatosis in a neonate. Arch Dermatol
2000;136:597-600
Vuoi citare questo contributo?